Ayrton, J-.P., Ho, C., Zhang, H., Chudasama, V., Frank S., Thomas, M. R.
Nanoscale, 2024, 16, 19881-19896
 Abstract
Abstract
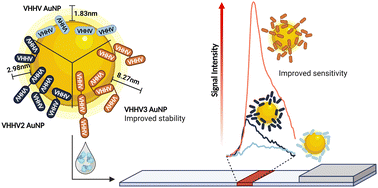
The ease of expression and engineering of single domain antibodies, known as nanobodies, make them attractive alternatives to conventional antibodies in point-of-care diagnostics such as lateral flow assays. In lateral flow assays, gold nanoparticle bioconjugates serve as labels which display affinity molecules on the gold surface. While examples of nanobody gold nanoparticle bioconjugates exist, few utilise the simple one-step approach of physisorption owing to undesirable nanoparticle aggregation and loss of functionality. Here we show that engineering nanobodies into multivalent structures can significantly enhance their functionality when physisorbed onto gold nanoparticles. This approach enables resulting bioconjugates to withstand multiple processing steps required for long-term nanoparticle storage within lateral flow assays. Specifically, we show that the trivalent version of VHHV nanobody (VHH3) against the S1 protein of SARS-CoV-2 can be immobilised onto gold nanoparticles through passive adsorption. Unlike its monovalent and bivalent nanobody counterparts, using VHHV3 preserves nanoparticle stability under salt stress, blocking, washing, and freeze-drying conditions while maintaining picomolar sensitivity to the S1 protein. We anticipate that this facile strategy is a significant advancement towards the integration of nanobodies in lateral flow assay development.
Shajan, I., Rochet, L. N. C., Tracey, S. R., Benazza, R., Jackowska, B., Hernandez-Alba, O., Cianférani, S., Scott, C. J., van Delft, F. L., Chudasama, V., Albada, B.
Bioconjugate Chem, 2024, 10, 1524–1531
 Abstract
Abstract
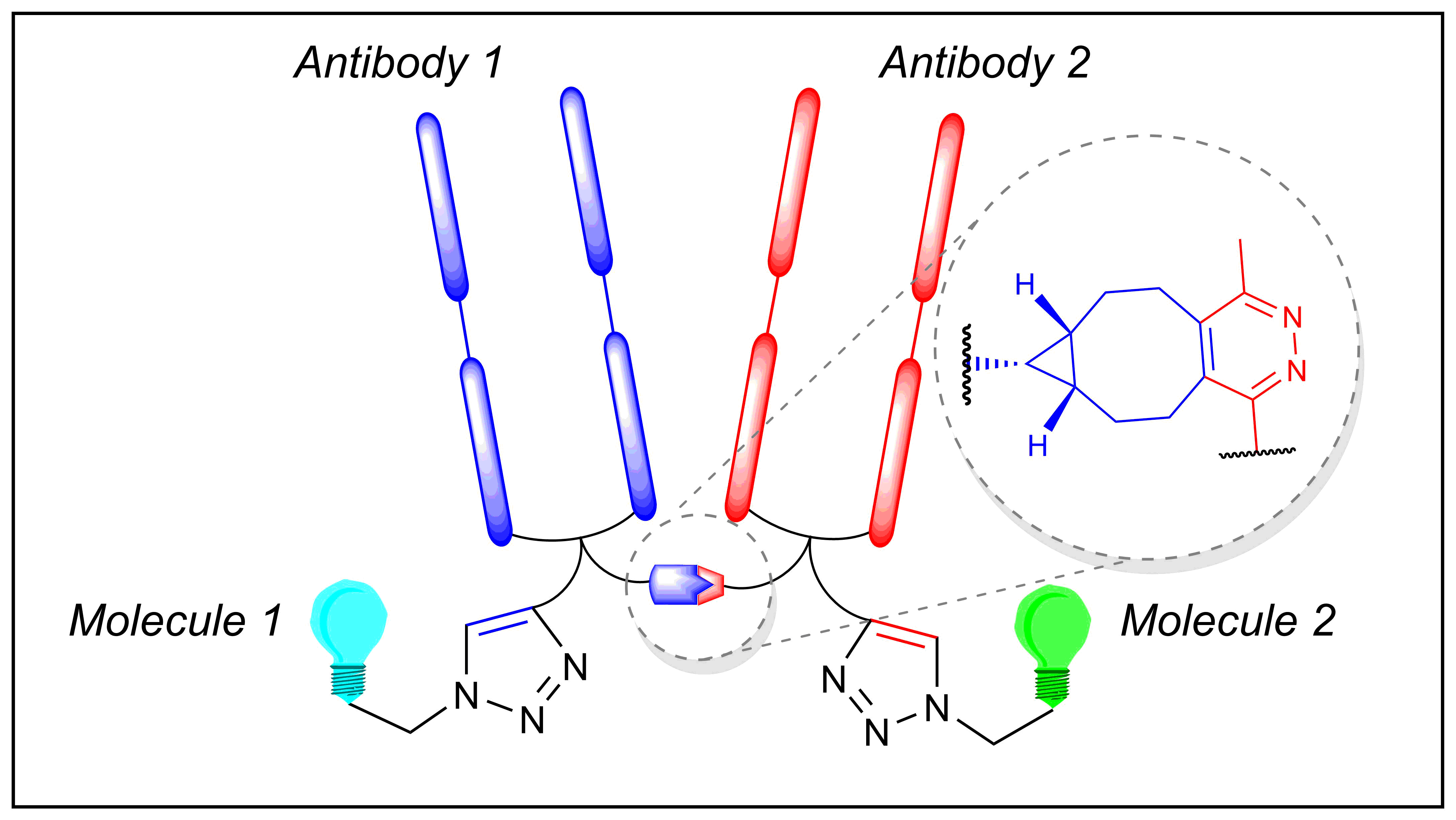
Redirecting T cells to tumor cells by bispecific antibodies is an effective approach to treat cancer, and T cell-dependent bispecific antibodies (TDBAs) are an emerging class of potent immunotherapeutic agents. By simultaneously targeting antigens on tumor cells and T cells, T cells are activated to kill tumor cells. Herein, we report a platform to generate a novel class of 2:1 structure of T cell-dependent bispecific antibody with bivalency for HER2 receptors on tumor cells and monovalency for CD3 receptors on T cells. For this, we use a biogenic inverse electron-demand Diels–Alder (IEDDA) click reaction on genetically encoded tyrosine residues to install one TCO handle on therapeutically approved antibody trastuzumab. Subsequent TCO-tetrazine click with a tetrazine-functionalized CD3-binding Fab yields a 2:1 HER2 × CD3 TDBA that exhibits a tumor-killing capability at picomolar concentrations. Monovalency toward the CD3 receptor on T cells can lower the chances of cytokine release syndrome, which is a common side effect of such agents. Our semisynthetic approach can generate highly potent TDBA constructs in a few chemoenzymatic and synthetic steps.
Yap, S. Y., Butcher, T., Spears, R. J., McMahon, C., Thanasi, I. A., Baker, J. R., Chudasama, V.*
Chem Sci, 2024, 15, 8557-8568
 Abstract
Abstract
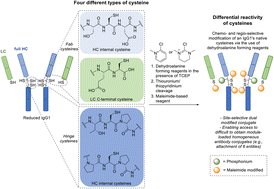
Protein modification has garnered increasing interest over the past few decades and has become an important tool in many aspects of chemical biology. In recent years, much effort has focused on site-selective modification strategies that generate more homogenous bioconjugates, and this is particularly so in the antibody modification space. Modifying native antibodies by targeting solvent-accessible cysteines liberated by interchain disulfide reduction is, perhaps, the predominant strategy for achieving more site-selectivity on an antibody scaffold. This is evidenced by numerous approved antibody therapeutics that have utilised cysteine-directed conjugation reagents and the plethora of methods/strategies focused on antibody cysteine modification. However, all of these methods have a common feature in that after the reduction of native solvent-accessible cystines, the liberated cysteines are all reacted in the same manner. Herein, we report the discovery and application of dehydroalanine forming reagents (including novel reagents) capable of regio- and chemo-selectively modifying these cysteines (differentially) on a clinically relevant antibody fragment and a full antibody. We discovered that these reagents could enable differential reactivity between light chain C-terminal cysteines, heavy chain hinge region cysteines (cysteines with an adjacent proline residue, Cys–Pro), and other heavy chain internal cysteines. This differential reactivity was also showcased on small molecules and on the peptide somatostatin. The application of these dehydroalanine forming reagents was exemplified in the preparation of a dually modified antibody fragment and full antibody. Additionally, we discovered that readily available amide coupling agents can be repurposed as dehydroalanine forming reagents, which could be of interest to the broader field of chemical biology.
Tapia, A. R., Abgottspon, F., Nilvebrant, J., Nygren, P.-Å., Ivetich, S. D., Hernandez, A. J. B., Thanasi, I. A., Szijj, P. A., Sekkat, G., Cuenot, F. M., Chudasama, V., Aceto, N., deMello, A. J., Richards, D. A.
Chem Sci, 2024, 15, 8982-8992
 Abstract
Abstract
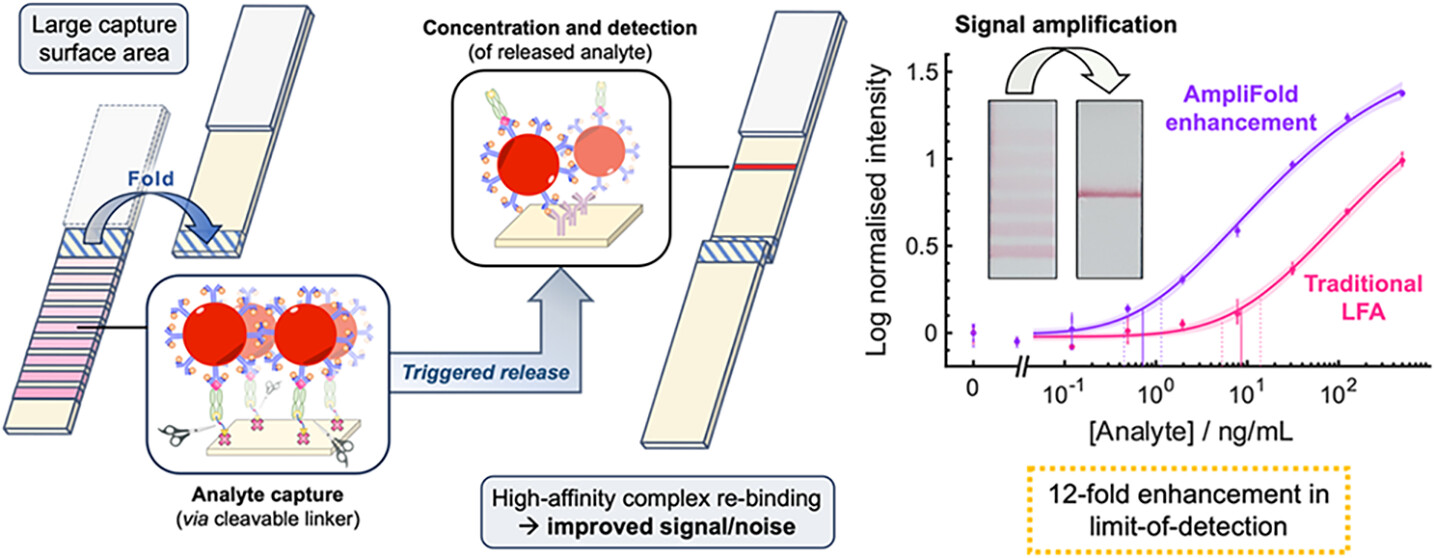
Affinity protein–oligonucleotide conjugates are increasingly being explored as diagnostic and therapeutic tools. Despite growing interest, these probes are typically constructed using outdated, non-selective chemistries, and little has been done to investigate how conjugation to oligonucleotides influences the function of affinity proteins. Herein, we report a novel site-selective conjugation method for furnishing affinity protein–oligonucleotide conjugates in a 93% yield within fifteen minutes. Using SPR, we explore how the choice of affinity protein, conjugation strategy, and DNA length impact target binding and reveal the deleterious effects of non-specific conjugation methods. Furthermore, we show that these adverse effects can be minimised by employing our site-selective conjugation strategy, leading to improved performance in an immuno-PCR assay. Finally, we investigate the interactions between affinity protein–oligonucleotide conjugates and live cells, demonstrating the benefits of site-selective conjugation. This work provides critical insight into the importance of conjugation strategy when constructing affinity protein–oligonucleotide conjugates.
Schauenburg, D., Gao, B., Rochet, L. N. C., Schüler, D., Coelho, J. A. S., Ng, D. Y. W., Chudasama, V., Kuan, S. L., Weil, T.
Angew Chem Int Ed, 2024, e202314143
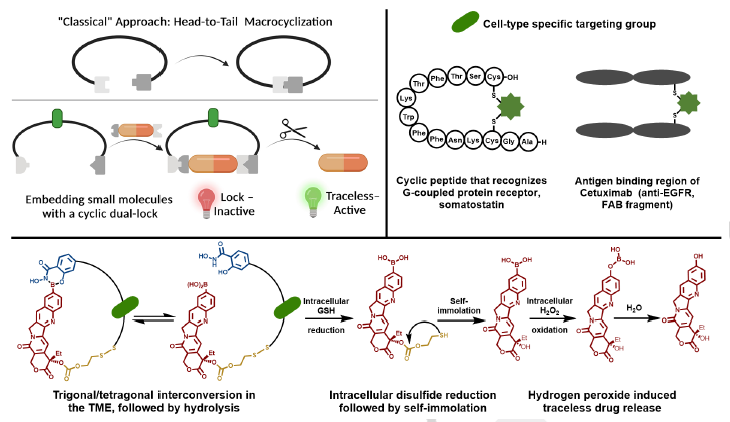
Abstract
Drug safety and efficacy due to premature release into the bloodstream and poor biodistribution remains a problem despite seminal advances in this area. To circumvent these limitations, we report drug cyclization based on dynamic covalent linkages to devise a dual lock for the small molecule anticancer drug, camptothecin (CPT). Drug activity is “locked” within the cyclic structure by the redox responsive disulfide and pH-responsive boronic acid-salicylhydroxamate and turns on only in the presence of acidic pH, reactive oxygen species and glutathione through traceless release. Notably, the dual-responsive CPT is more active (100-fold) than the non-cleavable (permanently closed) analogue. We further include a bioorthogonal handle in the backbone for functionalization to generate cyclic-locked, cell-targeting peptide- and protein-CPTs, for targeted delivery of the drug and traceless release in triple negative metastatic breast cancer cells to inhibit cell growth at low nanomolar concentrations.
Dinesen, A., Andersen, V. L., …, Chudasama, V., Baker, J. R., Gothelf, K. V., Wengel, J., Kjems, J., Howard, K. A.
Bioconjugate Chem, 2024, 35, 214-222
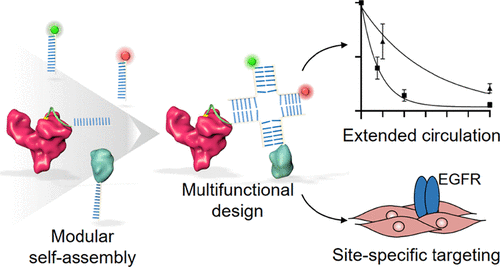
Abstract
Combinatorial properties such as long-circulation and site- and cell-specific engagement need to be built into the design of advanced drug delivery systems to maximize drug payload efficacy. This work introduces a four-stranded oligonucleotide Holliday Junction (HJ) motif bearing functional moieties covalently conjugated to recombinant human albumin (rHA) to give a “plug-and-play” rHA-HJ multifunctional biomolecular assembly with extended circulation. Electrophoretic gel-shift assays show successful functionalization and purity of the individual high-performance liquid chromatography-purified modules as well as efficient assembly of the rHA-HJ construct. Inclusion of an epidermal growth factor receptor (EGFR)-targeting nanobody module facilitates specific binding to EGFR-expressing cells resulting in approximately 150-fold increased fluorescence intensity determined by flow cytometric analysis compared to assemblies absent of nanobody inclusion. A cellular recycling assay demonstrated retained albumin-neonatal Fc receptor (FcRn) binding affinity and accompanying FcRn-driven cellular recycling. This translated to a 4-fold circulatory half-life extension (2.2 and 0.55 h, for the rHA-HJ and HJ, respectively) in a double transgenic humanized FcRn/albumin mouse. This work introduces a novel biomolecular albumin-nucleic acid construct with extended circulatory half-life and programmable multifunctionality due to its modular design.
Patel, M., Forte, N., Bishop, C. R., Porter, M. J., Dagwell, M., Karu,, K., Chudasama, V.*, Baker, J. R.
J Am Chem Soc, 2024, 146, 274–280

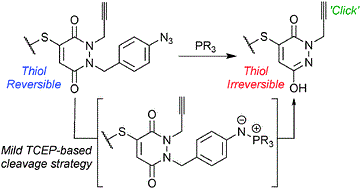
Abstract
Electron-poor aryl nitriles are promising reagents for bioconjugation due to their high electrophilicity and selectivity for reaction with thiols, albeit generally in a reversible manner. A transient species has previously been observed in such reactions, involving the addition of two thiols to the nitrile functional group, forming a tetrahedral amino dithioacetal (ADTA). In this work, the reaction of heteroaryl nitriles with bis-thiols is explored in an attempt to generate stable ADTAs, which could facilitate new bioconjugation protocols. By use of a 1,2-dithiol, or the incorporation of an electrophilic trap into the aryl nitrile design, the formation of stable products is achieved. The resultant “nitrile bis-thiol” (NBT) reaction is then explored in the context of protein modification, specifically to carry out antibody conjugation. By addition of these nitriles to the reduced disulfide bond of an antibody fragment, it is shown that, depending on the reagent design, cysteine-to-lysine transfer or disulfide bridged NBT products can be generated. Both represent site-selective conjugates and are shown to be stable when challenged with glutathione under physiological conditions and upon incubation in serum. Furthermore, the NBT reaction is tested in the more challenging context of a full antibody, and all four disulfide bonds are effectively modified by these new one-carbon bridging reagents. Overall, this reaction of heteroaryl-nitriles with bis-thiols is shown to be highly efficient and versatile, of tunable reversibility, and offers enticing prospects as a new addition to the toolbox of biocompatible “click”-type reactions.
Rochet, L. N. C., Bahou, C., Wojciechowski, J. P., Koutsopetras, I., Britton, P., Spears, R. J., Thanasi, I. A., Shao, B., Zhong, L., Bučar, D. K., Aliev, A. E., Porter, M. J., Stevens, M. M., Baker, J. R. Chudasama, V.*
Chem Sci, 2023, 14, 13743-13754
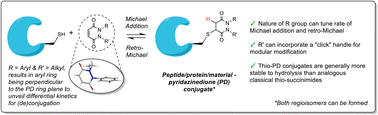
Abstract
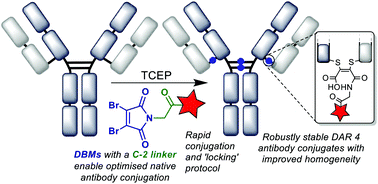
Reversible cysteine modification has been found to be a useful tool for a plethora of applications such as selective enzymatic inhibition, activity-based protein profiling and/or cargo release from a protein or a material. However, only a limited number of reagents display reliable dynamic/reversible thiol modification and, in most cases, many of these reagents suffer from issues of stability, a lack of modularity and/or poor rate tunability. In this work, we demonstrate the potential of pyridazinediones as novel reversible and tuneable covalent cysteine modifiers. We show that the electrophilicity of pyridazinediones correlates to the rates of the Michael addition and retro-Michael deconjugation reactions, demonstrating that pyridazinediones provide an enticing platform for readily tuneable and reversible thiol addition/release. We explore the regioselectivity of the novel reaction and unveil the reason for the fundamental increased reactivity of aryl bearing pyridazinediones by using DFT calculations and corroborating findings with SCXRD. We also applied this fundamental discovery to making more rapid disulfide rebridging agents in related work. We finally provide the groundwork for potential applications in various areas with exemplification using readily functionalised “clickable” pyridazinediones on clinically relevant cysteine and disulfide conjugated proteins, as well as on a hydrogel material.
Shajan, I., Rochet, L. N. C., Tracey, S. R., Jackowska, B., Benazza, R., Hernandez-Alba, O., Cianférani, S., Scott, C. J., van Delft, F. L., Chudasama, V.*, Albada, B.
Bioconjugate Chem, 2023, 34, 2215–2220
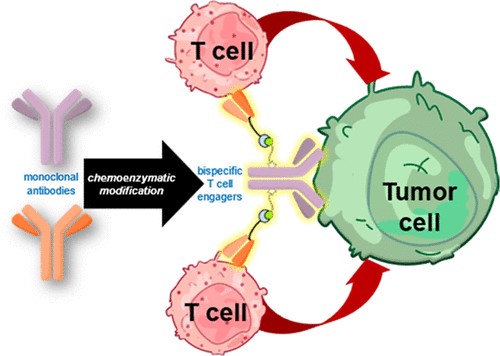
Abstract
Bispecific antibodies as T cell engagers designed to display binding capabilities to both tumor-associated antigens and antigens on T cells are considered promising agents in the fight against cancer. Even though chemical strategies to develop such constructs have emerged, a method that readily converts a therapeutically applied antibody into a bispecific construct by a fully non-genetic process is not yet available. Herein, we report the application of a biogenic, tyrosine-based click reaction utilizing chemoenzymatic modifications of native IgG1 antibodies to generate a synthetic bispecific antibody construct that exhibits tumor-killing capability at picomolar concentrations. Control experiments revealed that a covalent linkage of the different components is required for the observed biological activities. In view of the highly potent nature of the constructs and the modular approach that relies on convenient synthetic methods utilizing therapeutically approved biomolecules, our method expedites the production of potent bispecific antibody constructs with tunable cell killing efficacy with significant impact on therapeutic properties.
Bahou, C., Spears, R. J., Rosales, A. M. R., Rochet, L. N. C., Barber, L. J., Stankevich, K. S., Miranda, J. F., Butcher, T. C., Kerrigan, A. M., Lazarov, V. K., Grey, W., Chudasama, V.*, Spicer, C. D.
Biomacromolecules, 2023, 24, 4646-4652.
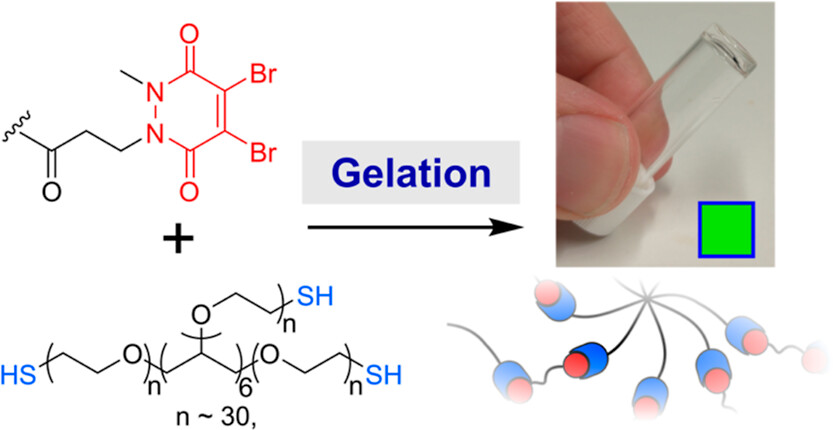
Abstract
Thiol-reactive Michael acceptors are commonly used for the formation of chemically cross-linked hydrogels. In this paper, we address the drawbacks of many Michael acceptors by introducing pyridazinediones as new cross-linking agents. Through the use of pyridazinediones and their mono- or dibrominated analogues, we show that the mechanical strength, swelling ratio, and rate of gelation can all be controlled in a pH-sensitive manner. Moreover, we demonstrate that the degradation of pyridazinedione-gels can be induced by the addition of thiols, thus providing a route to responsive or dynamic gels, and that monobromo-pyridazinedione gels are able to support the proliferation of human cells. We anticipate that our results will provide a valuable and complementary addition to the existing toolkit of cross-linking agents, allowing researchers to tune and rationally design the properties of biomedical hydrogels.
Yanbo, Z., Chudasama, V.*, Baker, J. R.
ChemBioChem, 2023, 24, e202300356.
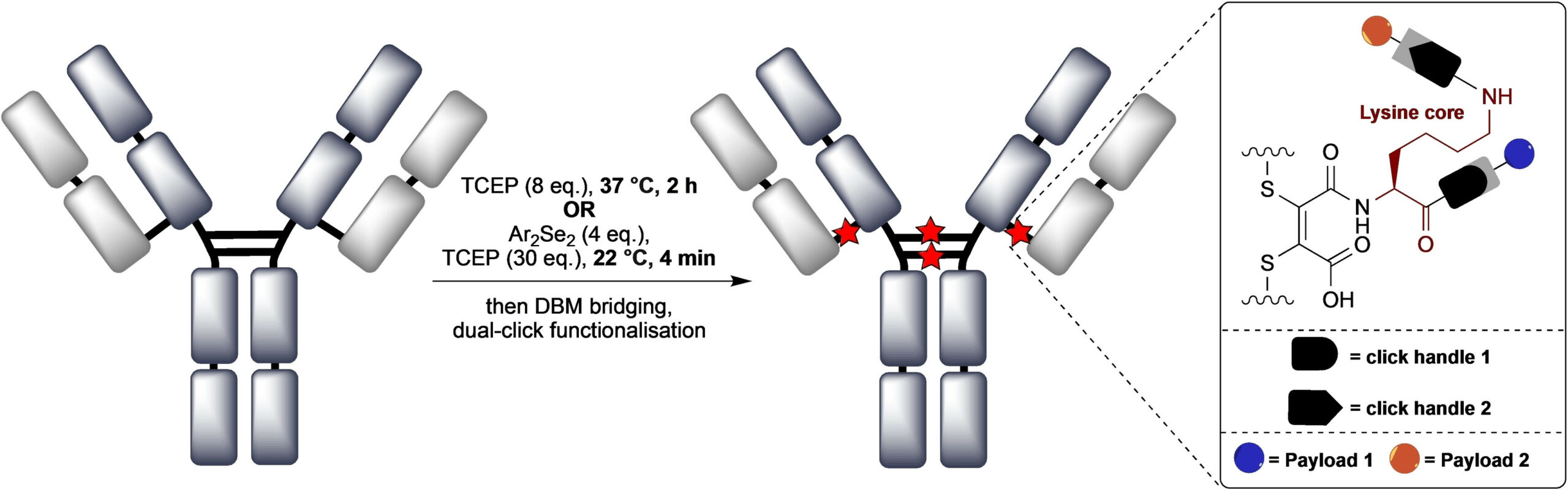
Abstract
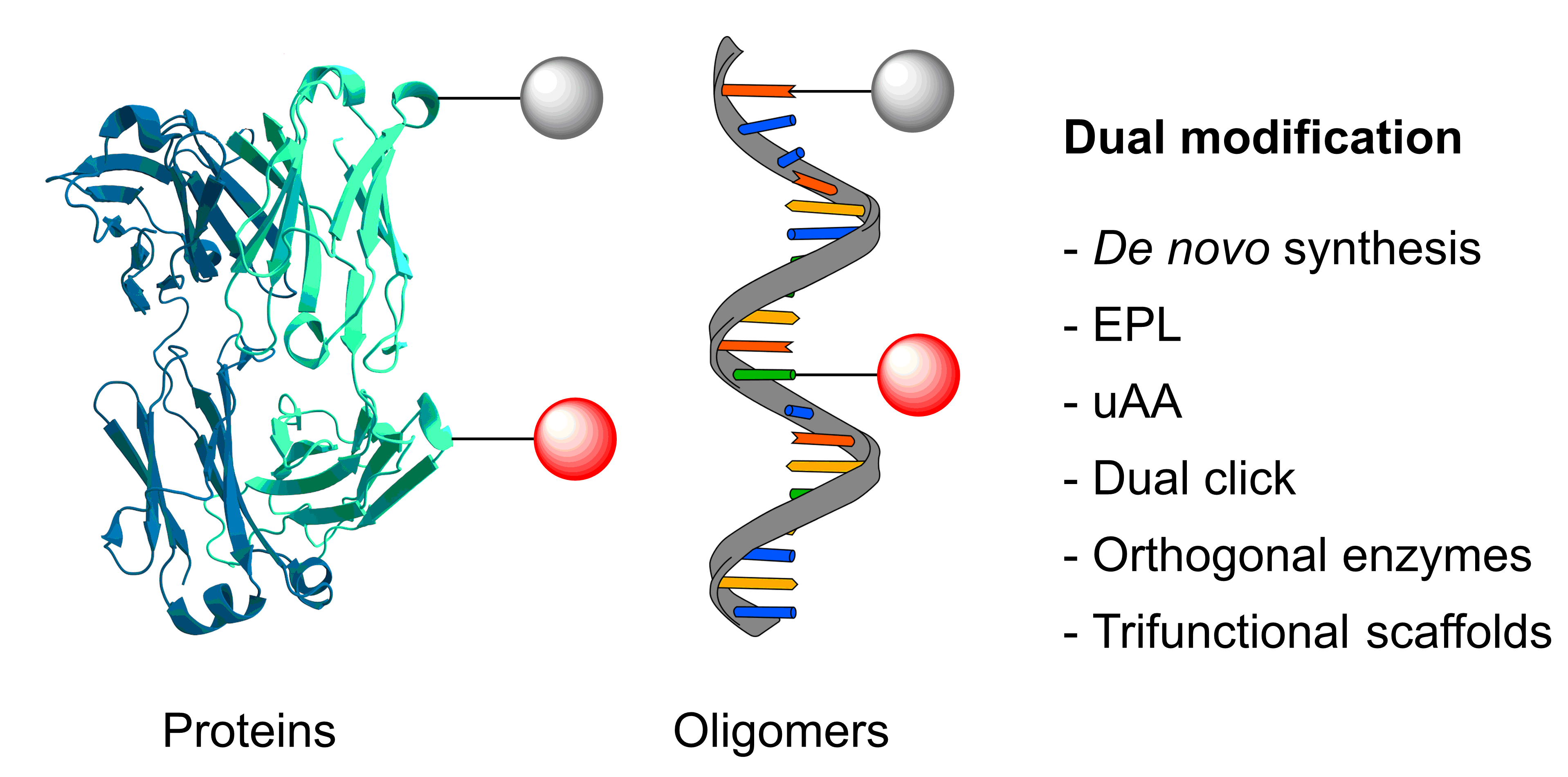
We describe the synthesis and application of a selection of trifunctional reagents for the dual-modality modification of native, solvent accessible disulfide bonds in trastuzumab. The reagents were developed from the dibromomaleimide (DBM) platform with two orthogonal clickable functional groups built around a lysine core. We also describe the development of an aryl diselenide additive which enables antibody disulfide reduction in 4 minutes and a rapid overall reduction-bridging-double click sequence.
Szijj, P. A., Gray, M. A., Ribi, M. K., Bahou, C., Nogueira, J. C. F., Bertozzi, C. R., Chudasama, V.*
Nature Chem, 2023, 15, 1636-1647
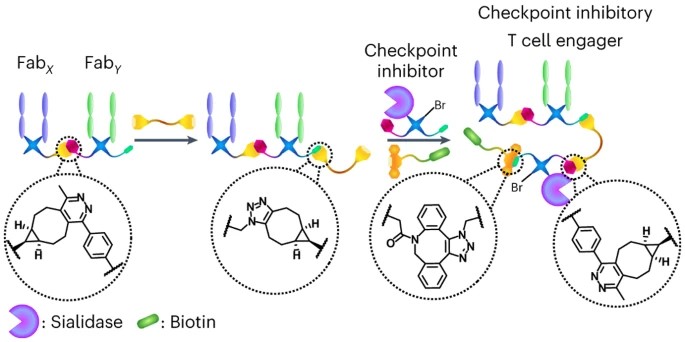
Abstract
Bispecific T cell engagers (BiTEs), a subset of bispecific antibodies (bsAbs), can promote a targeted cancer cell’s death by bringing it close to a cytotoxic T cell. Checkpoint inhibitory T cell engagers (CiTEs) comprise a BiTE core with an added immunomodulatory protein, which serves to reverse cancer-cell immune-dampening strategies, improving efficacy. So far, protein engineering has been the main approach to generate bsAbs and CiTEs, but improved chemical methods for their generation have recently been developed. Homogeneous fragment-based bsAbs constructed from fragment antigen-binding regions (Fabs) can be generated using click chemistry. Here we describe a chemical method to generate biotin-functionalized three-protein conjugates, which include two CiTE molecules, one containing an anti-PD-1 Fab and the other containing an immunomodulatory enzyme, Salmonella typhimurium sialidase. The CiTEs’ efficacy was shown to be superior to that of the simpler BiTE scaffold, with the sialidase-containing CiTE inducing substantially enhanced T cell-mediated cytotoxicity in vitro. The chemical method described here, more generally, enables the generation of multi-protein constructs with further biological applications.
Spears, R. J., Chudasama, V.*
Curr Opin Chem Bio, 2023, 75, 102306.
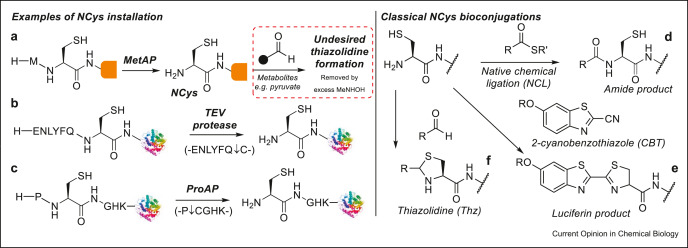
Abstract
Advances in the site-specific chemical modification of proteins, also referred to as protein bioconjugation, have proved instrumental in revolutionary approaches to designing new protein-based therapeutics. Of the sites available for protein modification, cysteine residues or the termini of proteins have proved especially popular owing to their favorable properties for site-specific modification. Strategies that, therefore, specifically target cysteine at the termini offer a combination of these favorable properties of cysteine and termini bioconjugation. In this review, we discuss these strategies with a particular focus on those reported recently and provide our opinion on the future direction of the field.
Malde, R., Richards, D. A., Salerno, L., Zhao, Y., Karu, K., Chudasama, V.*, Baker, J. R.
Eur J Org Chem, 2023, 26, e202201372.
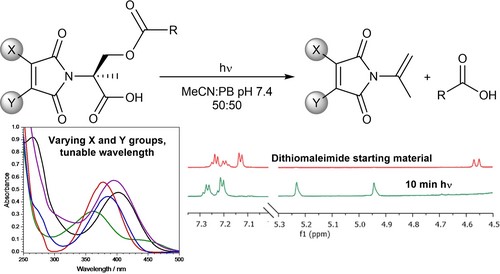
Abstract
Herein we report on photodecarboxylations of various substituted maleimides, resulting in an elimination reaction. Furthermore, we establish facile wavelength tunability through modulation of the maleimide double bond substituents. We envisage that these versatile reagents, which are readily constructed and diversified by nucleophilic substitution reactions on bromomaleimides, will offer new opportunities for triggered photorelease.
Creamer, A., …, Rochet, L. N. C., Thanasi, I. A., …, Chudasama, V., Fenaroli, F., Stevens, M. M.
Adv Materials, 2023, 2300413.
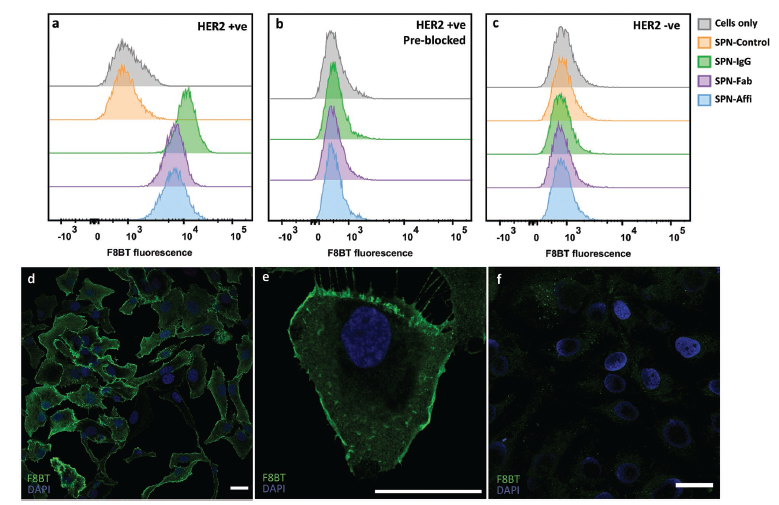
Abstract
Semiconducting polymer nanoparticles (SPNs) have been explored for applications in cancer theranostics because of their high absorption coefficients, photostability and biocompatibility. However, SPNs are susceptible to aggregation and protein fouling in physiological conditions, which can be detrimental for in vivo applications. Here, we describe a method for achieving colloidally stable and low-fouling SPNs by grafting PEG onto the backbone of the fluorescent semiconducting polymer, poly(9,9′-dioctylfluorene-5-fluoro-2,1,3-benzothiadiazole) (F8BT-F), in a simple one-step substitution reaction, post-polymerisation. Further, by utilising azide-functionalised PEG we site-specifically “click” anti-HER2 antibodies, Fab fragments, or affibodies onto the SPN surface, which allows the functionalised SPNs to specifically target HER2-positive cancer cells. In vivo, our PEGylated SPNs were found to have excellent circulation efficiencies in zebrafish embryos for up to seven days post-injection. SPNs functionalised with affibodies were then shown to be able to target HER2 expressing cancer cells in a zebrafish xenograft model. The covalent PEGylated SPN system described herein shows great potential for cancer theranostics.
Thoreau, F., Rochet, L. N. C., Baker, J. R., Chudasama, V.*
Chem Sci, 2023, 14, 3752-3762.
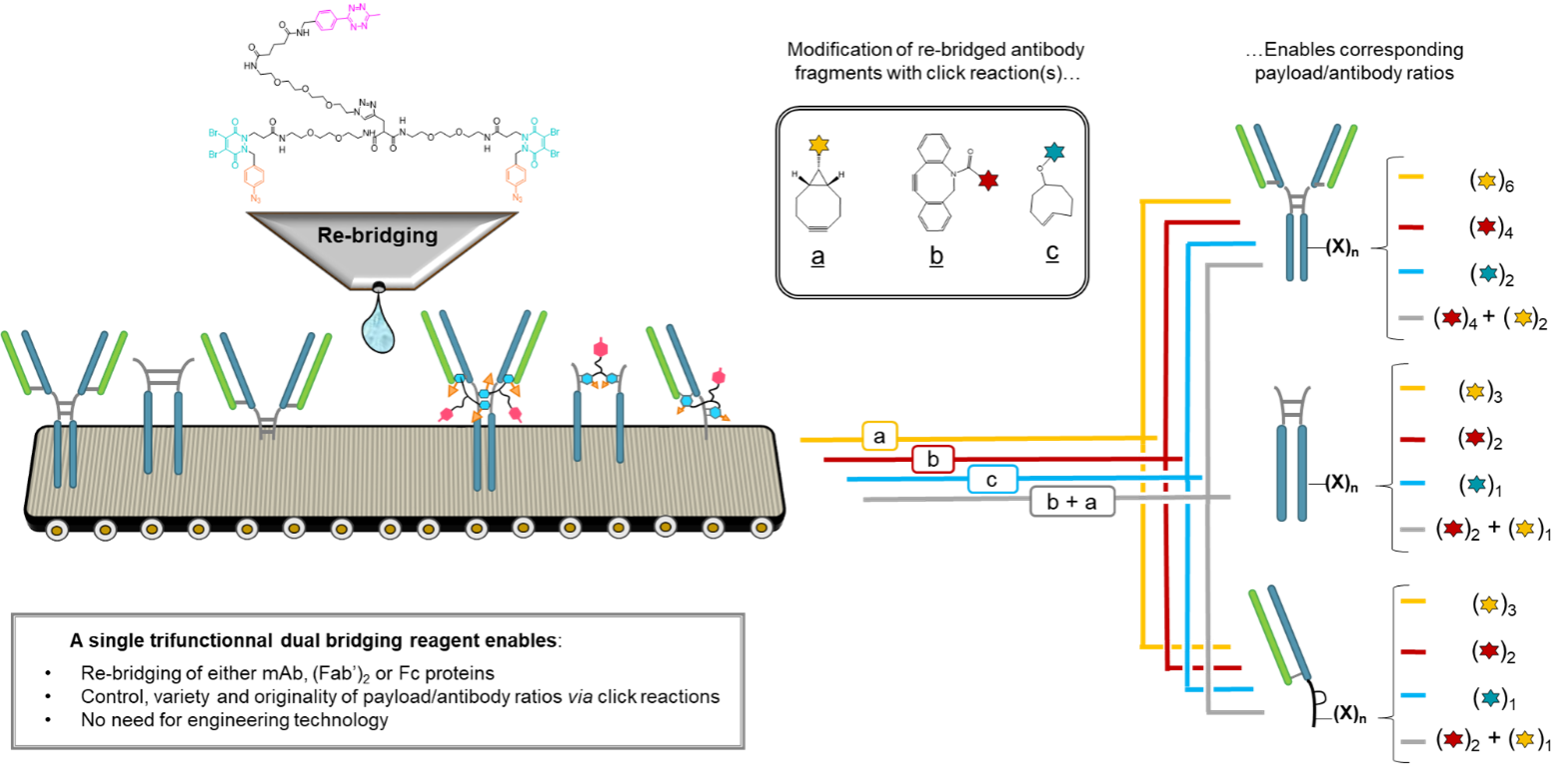 Abstract
Abstract
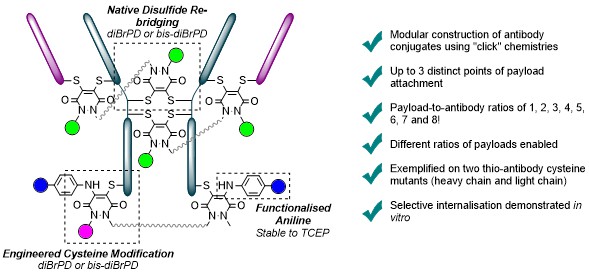
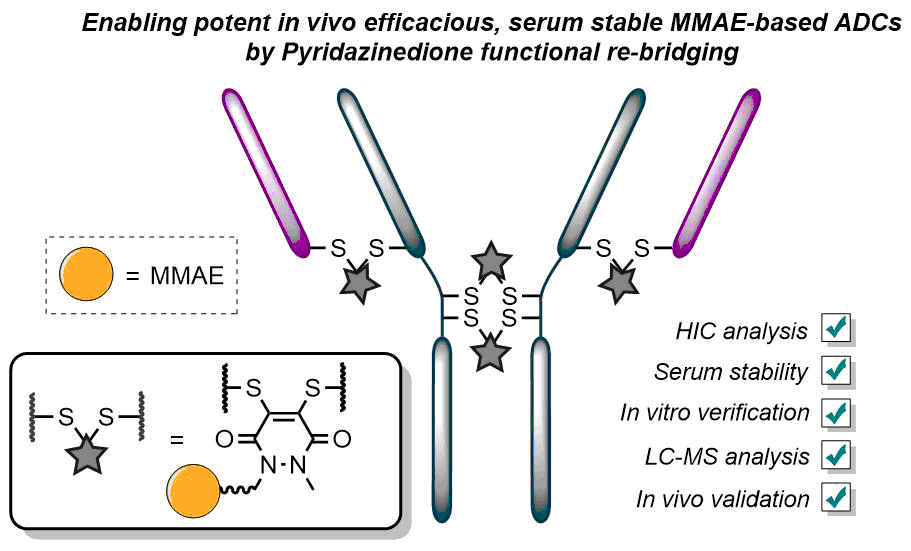
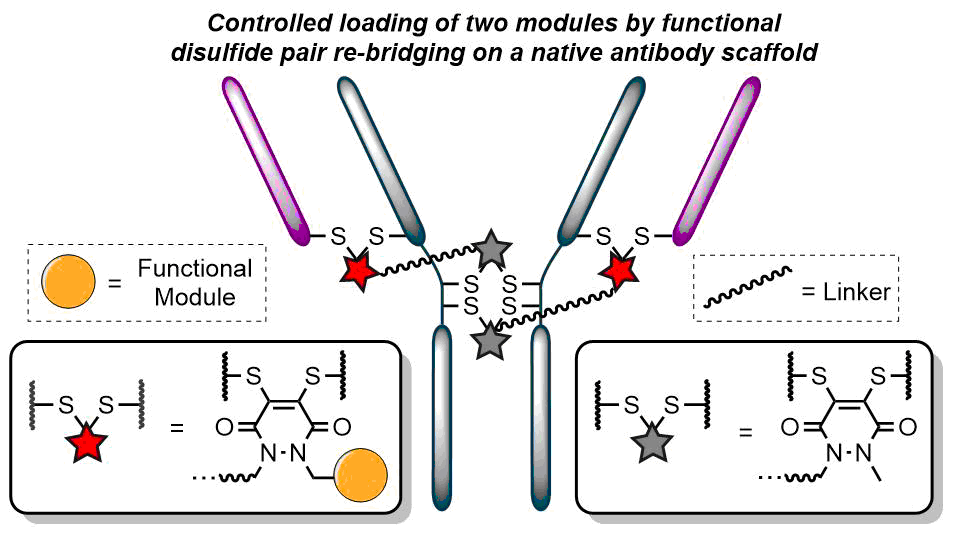
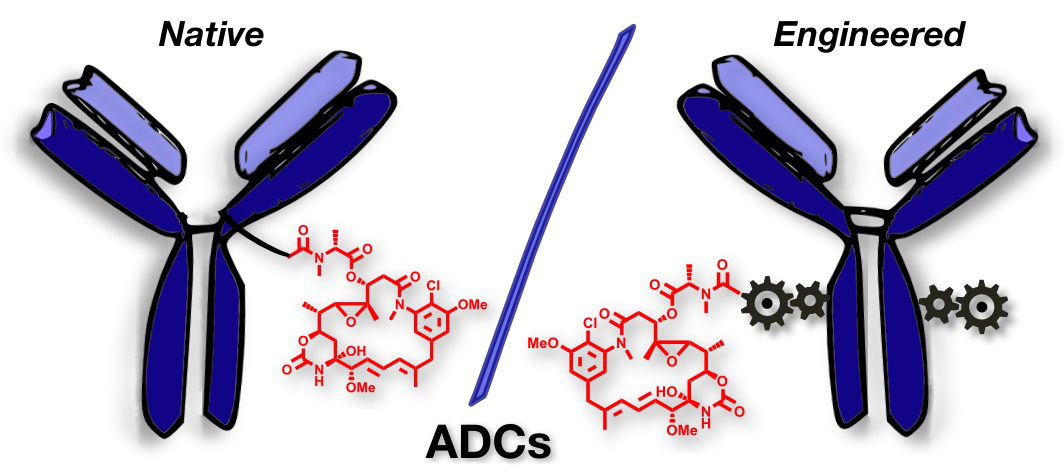
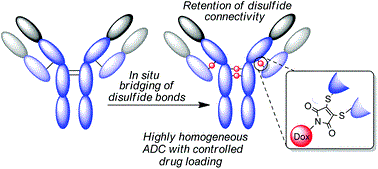
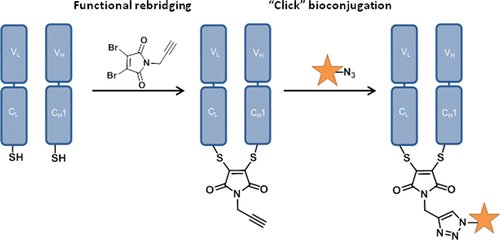
Either as full IgGs or as fragments (Fabs, Fc, etc.), antibodies have received tremendous attention in the development of new therapeutics such as antibody–drug conjugates (ADCs). The production of ADCs involves the grafting of active payloads onto an antibody, which is generally enabled by the site-selective modification of native or engineered antibodies via chemical or enzymatic methods. Whatever method is employed, controlling the payload–antibody ratio (PAR) is a challenge in terms of multiple aspects including: (i) obtaining homogeneous protein conjugates; (ii) obtaining unusual PARs (PAR is rarely other than 2, 4 or 8); (iii) using a single method to access a range of different PARs; (iv) applicability to various antibody formats; and (v) flexibility for the production of heterofunctional antibody-conjugates (e.g. attachment of multiple types of payloads). In this article, we report a single pyridazinedione-based trifunctional dual bridging linker that enables, in a two-step procedure (re-bridging/click), the generation of either mAb-, Fab′-, or Fc-conjugates from native mAb, (Fab′)2 or Fc formats, respectively. Fc and (Fab′)2 formats were generated via enzymatic digestion of native mAbs. Whilst the same reduction and re-bridging protocols were applied to all three of the protein formats, the subsequent click reaction(s) employed to graft payload(s) drove the generation of a range of PARs, including heterofunctional PARs. As such, exploiting click reactivity and/or orthogonality afforded mAb-conjugates with PARs of 6, 4, 2 or 4 + 2, and Fab′- and Fc-conjugates with a PAR of 3, 2, 1 or 2 + 1 on-demand. We believe that the homogeneity, novelty and variety in accessible PARs, as well as the applicability to various antibody-conjugate formats enabled by our non-recombinant method could be a suitable tool for antibody–drug conjugates optimisation (optimal PAR value, optimal payloads combination) and boost the development of new antibody therapeutics (Fab′- and Fc-conjugates).
Thoreau, F., Szijj, P. A., Greene, M. K., Rochet, L. N. C., Thanasi, I. A., Blayney, J. K., Maruani, A., Baker, J. R., Scott, C. J., Chudasama, V.*
ACS Cent Sci, 2023, 9, 476–487.
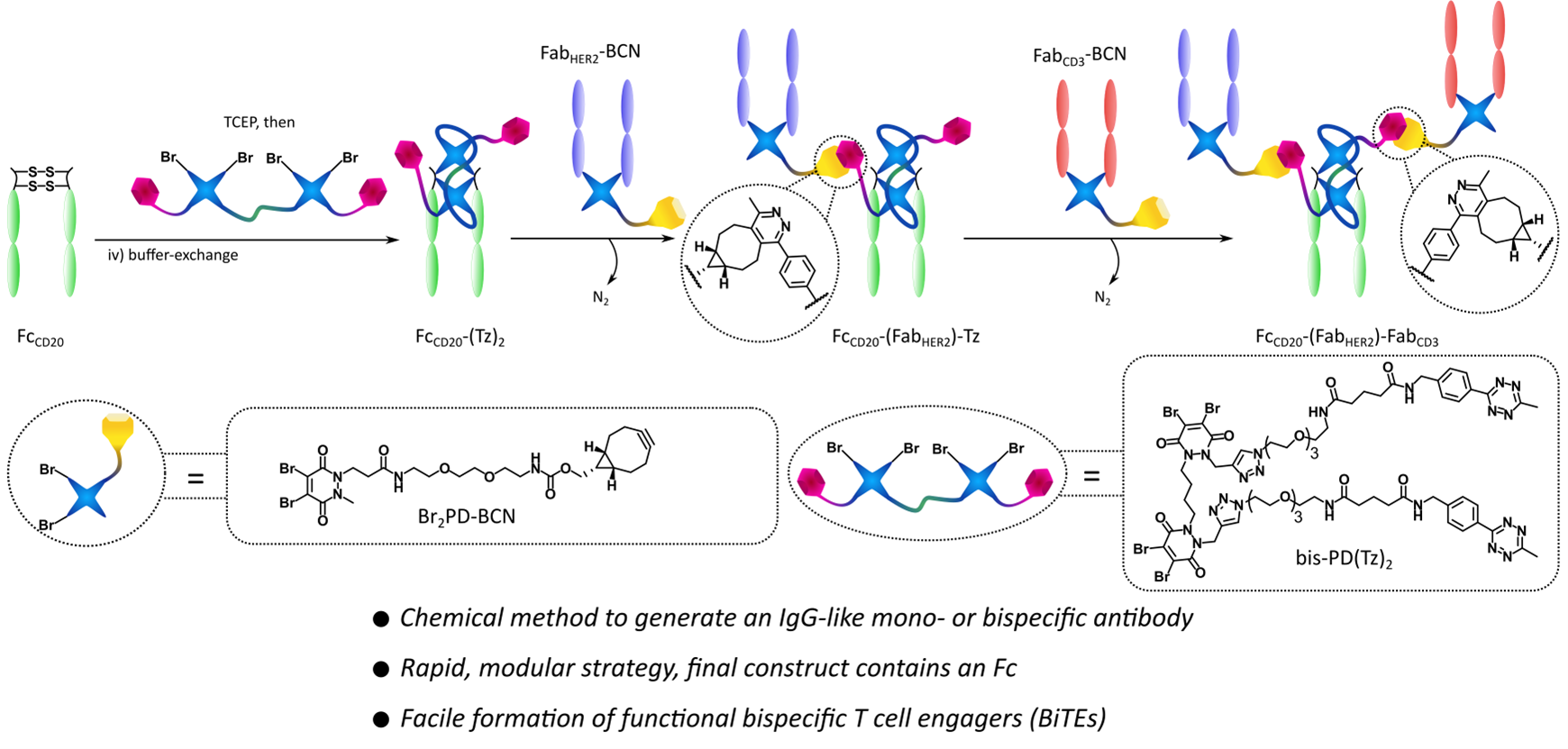 Abstract
Abstract
In recent years there has been rising interest in the field of protein–protein conjugation, especially related to bispecific antibodies (bsAbs) and their therapeutic applications. These constructs contain two paratopes capable of binding two distinct epitopes on target molecules and are thus able to perform complex biological functions (mechanisms of action) not available to monospecific mAbs. Traditionally these bsAbs have been constructed through protein engineering, but recently chemical methods for their construction have started to (re)emerge. While these have been shown to offer increased modularity, speed, and for some methods even the inherent capacity for further functionalization (e.g., with small molecule cargo), most of these approaches lacked the ability to include a fragment crystallizable (Fc) modality. The Fc component of IgG antibodies offers effector function and increased half-life. Here we report a first-in-class disulfide rebridging and click-chemistry-based method for the generation of Fc-containing, IgG-like mono- and bispecific antibodies. These are in the FcZ-(FabX)-FabY format, i.e., two distinct Fabs and an Fc, potentially all from different antibodies, attached in a homogeneous and covalent manner. We have dubbed these molecules synthetic antibodies (SynAbs). We have constructed a T cell-engager (TCE) SynAb, FcCD20-(FabHER2)-FabCD3, and have confirmed that it exhibits the expected biological functions, including the ability to kill HER2+ target cells in a coculture assay with T cells.
2022
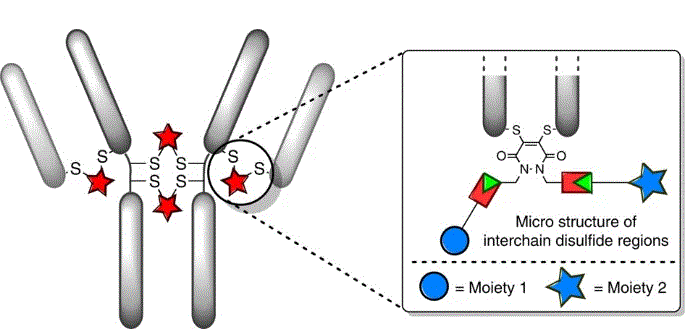
Chrzastek, A., Thanasi, I. A., Irving, J. A,, Chudasama, V.*, Baker, J. R.
Chem Sci, 2022, 13, 11533-11539.
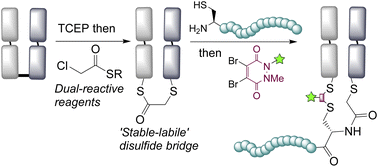 Abstract
Abstract
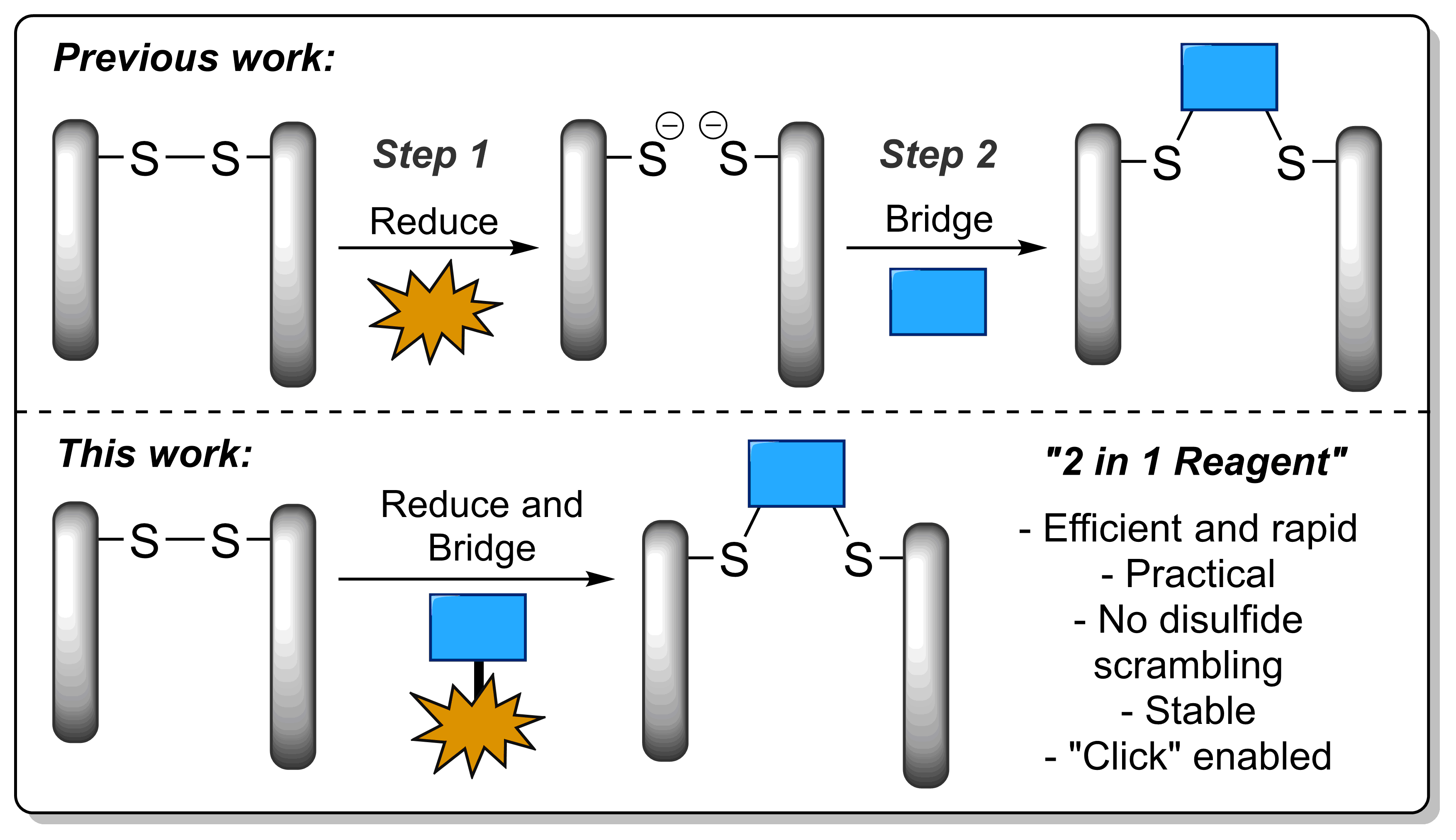
Disulfide bridging, also known as disulfide stapling, is a powerful strategy for the construction of site-selective protein bioconjugates. Here we describe the first examples of a new class of such reagents, containing a ‘stable-labile’ design. These dual-reactive reagents are designed to form a stable bond to one cysteine and a labile bond to the second; resulting in a robust attachment to the protein with one end of the bridge, whilst the other end serves as a reactive handle for subsequent bioconjugation. By incorporating thioesters into these bridges, we demonstrate that they are primed for native chemical ligation (NCL) with N-terminal cysteines; offering an alternative to the requirement for C-terminal thioesters for use in such ligations. Alternatively, the use of hydrazine as the ligating nucleophile enables a separate cargo to be attached to each cysteine residue, which are exploited to insert variably cleavable linkers. These methodologies are demonstrated on an antibody fragment, and serve to expand the scope of disulfide bridging strategies whilst offering a convenient route to the construction of multifunctional antibody fragment conjugates.
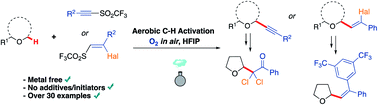
Ahmed, N., Spears, R. J., Sheppard, T., Chudasama, V.*
Chem Sci, 2022, 13, 8626-8633.
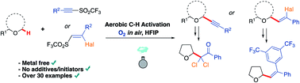 Abstract
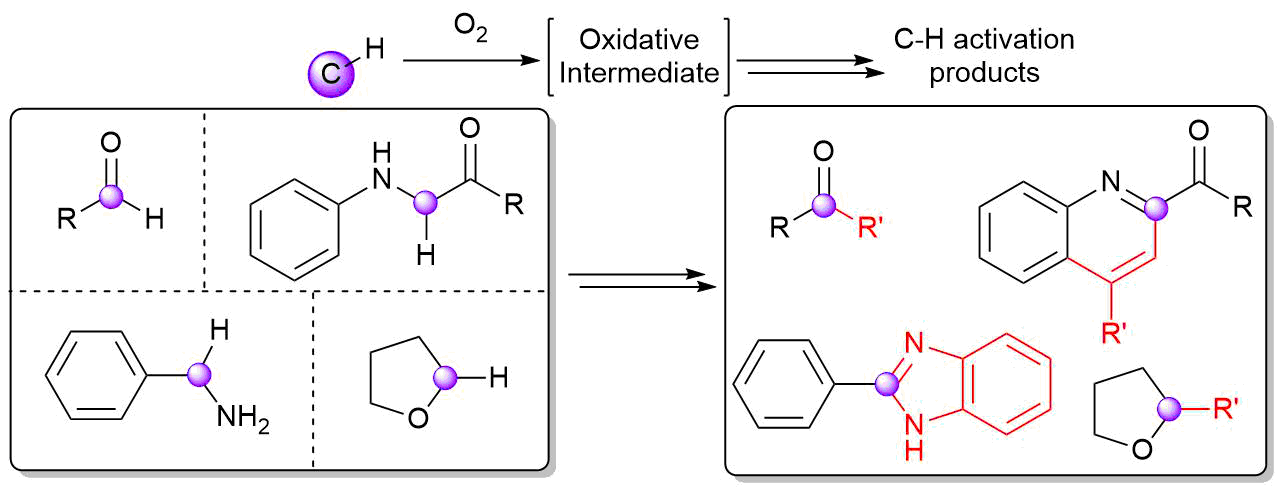
AbstractWith an ever-growing emphasis on sustainable synthesis, aerobic C–H activation (the use of oxygen in air to activate C–H bonds) represents a highly attractive conduit for the development of novel synthetic methodologies. Herein, we report the air mediated functionalisation of various saturated heterocycles and ethers via aerobically generated radical intermediates to form new C–C bonds using acetylenic and vinyl triflones as radical acceptors. This enables access to a variety of acetylenic and vinyl substituted saturated heterocycles that are rich in synthetic value. Mechanistic studies and control reactions support an aerobic radical-based C–H activation mechanism.
Gutiérrez-Fernández, J., Javaid, F., Rossi, G., Chudasama, V., Greenwood, J., Moss, S. E., Luecke, H.
Acta Cryst, 2022, 8, 725-734.
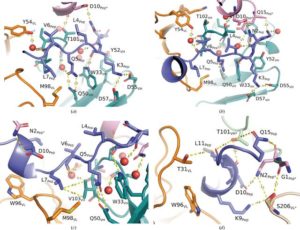 Abstract
AbstractThe formation of new dysfunctional blood vessels is a crucial stage in the development of various conditions such as macular degeneration, diabetes, cardiovascular disease, neurological disease and inflammatory disorders, as well as during tumor growth, eventually contributing to metastasis. An important factor involved in pathogenic angiogenesis is leucine-rich α-2-glycoprotein 1 (LRG1), the antibody blockade of which has been shown to lead to a reduction in both choroidal neovascularization and tumor growth in mouse models. In this work, the structural interactions between the LRG1 epitope and the Fab fragment of Magacizumab, a humanized function-blocking IgG4 against LRG1, are analysed, determining its specific binding mode and the key residues involved in LRG1 recognition. Based on these structural findings, a series of mutations are suggested that could be introduced into Magacizumab to increase its affinity for LRG1, as well as a model of the entire Fab–LRG1 complex that could enlighten new strategies to enhance affinity, consequently leading towards an even more efficient therapeutic.
Spears, R., Chrzastek, A., Yap, S., Karu, K., Aliev, A., Baker, J. R., Chudasama, V.*
Chem Commun, 2022, 58, 5359-5362.
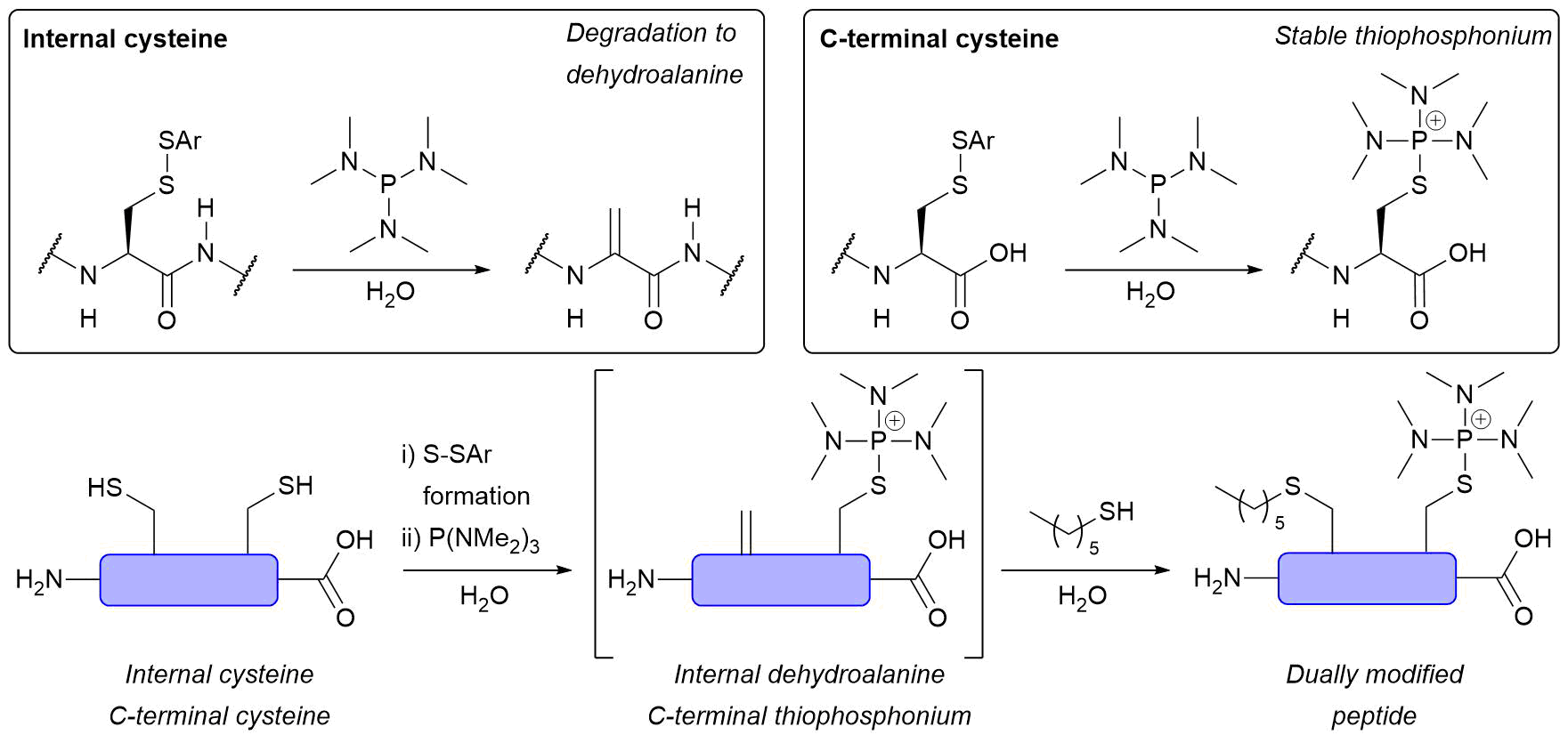 Abstract
AbstractHerein we report a fundamental discovery on the use of tris(dialkylamino)phosphine reagents for peptide and protein modification. We discovered that c terminal thiophosphonium species, which are uniquely stable, could be selectively and rapidly generated from their disulfide counterparts. In sharp and direct contrast, internal thiophosphonium species rapidly degrade to dha. We demonstrate this remarkable chemoselectivity on a bis-cysteine model peptide, and the formation of a stable c-terminal-thiophosphonium adduct on an antibody fragment, as well as characterise the species in various small molecule/peptide studies.
Bahou, C., Chudasama, V.*
Org Biomol Chem, 2022, 20, 5879-5890.
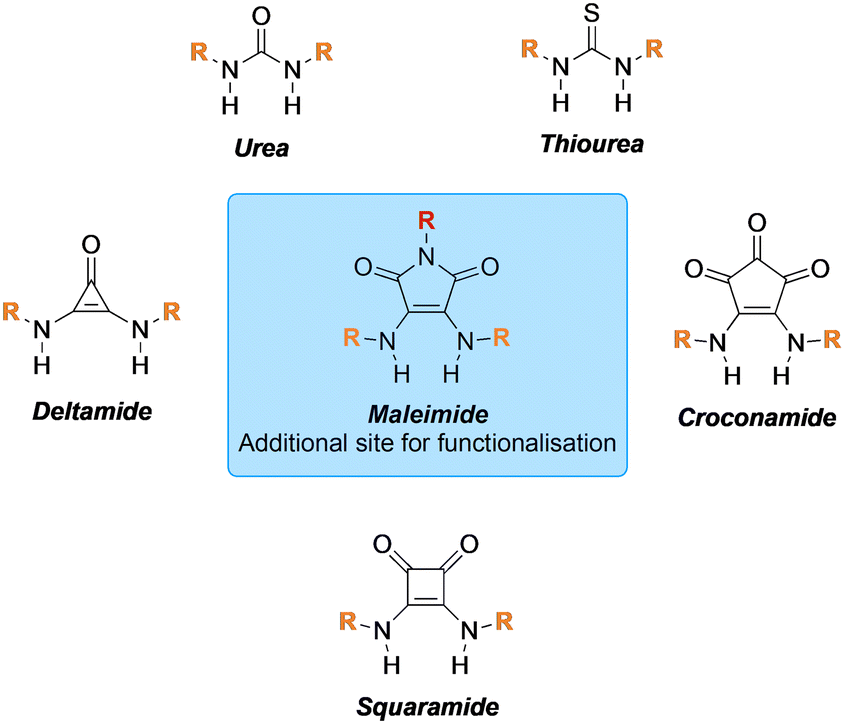
AbstractTools that facilitate the chemical modification of peptides and proteins are gaining an increasing amount of interest across many avenues of chemical biology as they enable a plethora of therapeutic, imaging and diagnostic applications. Cysteine residues and disulfide bonds have been highlighted as appealing targets for modification due to the highly homogenous nature of the products that can be formed through their site-selective modification. Amongst the reagents available for the site-selective modification of cysteine(s)/disulfide(s), pyridazinediones (PDs) have played a particularly important and enabling role. In this review, we outline the unique chemical features that make PDs especially well-suited to cysteine/disulfide modification on a wide variety of proteins and peptides, as well as provide context as to the problems solved (and applications enabled) by this technology.
Dinesen, A., Winther, A., Wall, A., Marcher, A., Palmfeldt, J., Chudasama, V., Wengel, J., Gothlef, K. V., Baker, J. R., Howard, K. A.
Bioconjugate Chem, 2022, 33, 333–342.
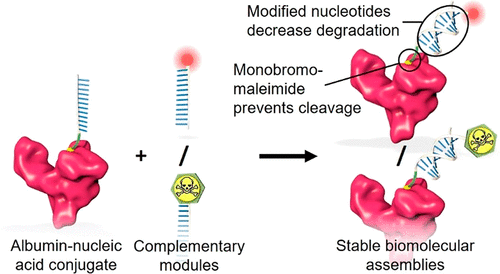
AbstractAlbumin-nucleic acid biomolecular drug designs offer modular multifunctionalization and extended circulatory half-life. However, stability issues associated with conventional DNA nucleotides and maleimide bioconjugation chemistries limit the clinical potential. This work aims to improve the stability of this thiol conjugation and nucleic acid assembly by employing a fast-hydrolyzing monobromomaleimide (MBM) linker and nuclease-resistant nucleotide analogues, respectively. The biomolecular constructs were formed by site-selective conjugation of a 12-mer oligonucleotide to cysteine 34 (Cys34) of recombinant human albumin (rHA), followed by annealing of functionalized complementary strands bearing either a fluorophore or the cytotoxic drug monomethyl auristatin E (MMAE). Formation of conjugates and assemblies was confirmed by gel shift analysis and mass spectrometry, followed by investigation of serum stability, neonatal Fc receptor (FcRn)-mediated cellular recycling, and cancer cell killing. The MBM linker afforded rapid conjugation to rHA and remained stable during hydrolysis. The albumin-nucleic acid biomolecular assembly composed of stabilized oligonucleotides exhibited high serum stability and retained FcRn engagement mediating FcRn-mediated cellular recycling. The MMAE-containing assembly exhibited cytotoxicity in the human MIA PaCa-2 pancreatic cancer cell line with an IC50 of 342 nM, triggered by drug release from breakdown of an acid-labile linker. In summary, this work presents rHA-nucleic acid module-based assemblies with improved stability and retained module functionality that further promotes the drug delivery potential of this biomolecular platform.
Maldi, R., Parkes, M. A., Staniforth, M., Woolley, J. M., Stavros, V. G., Chudasama, V.*, Fielding, H. H., Baker J. R.
Chem Sci, 2022, 13, 2909-2918.
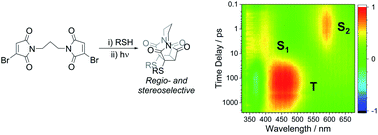
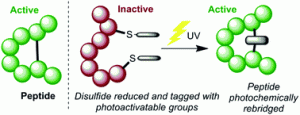
Abstract Thiomaleimides undergo efficient intermolecular [2 + 2] photocycloaddition reactions and offer applications from photochemical peptide stapling to polymer crosslinking; however, the reactions are limited to the formation of the exo head-to-head isomers. Herein, we present an intramolecular variation which completely reverses the stereochemical outcome of this photoreaction, quantitatively generating endo adducts which minimise the structural disturbance of the disulfide staple and afford a 10-fold increase in quantum yield. We demonstrate the application of this reaction on a protein scaffold, using light to confer thiol stability to an antibody fragment conjugate. To understand more about this intriguing class of [2 + 2] photocycloadditions, we have used transient absorption spectroscopy (electronic and vibrational) to study the excited states involved. The initially formed S2 (π1π*) excited state is observed to decay to the S1 (n1π*) state before intersystem crossing to a triplet state. An accelerated intramolecular C–C bond formation provides evidence to explain the increased efficiency of the reaction, and the impact of the various excited states on the carbonyl vibrational modes is discussed.
Thoreau, F., Chudasama, V.*
RSC Chem Bio, 2022, 3, 140-169.

AbstractIn the last two decades, immunotherapy has established itself as one of the leading strategy for cancer treatment, as illustrated by the exponentially growing number of related clinical trials. This trend was, in part, prompted by the clinical success of both immune checkpoint modulation and immune cell engagement, to restore and/or stimulate the patient’s immune system’s ability to fight the disease. These strategies were sustained by progress in bispecific antibody production. However, despite the decisive progress made in the treatment of cancer, toxicity and resistance are still observed in some cases. In this review, we intitally provide an overview of the monoclonal and bispecific antibodies developed with the objective to restore immune system functions to treat cancer (cancer immunotherapy), either being through immune checkpoint modulation, immune cell engagement or a combination of both. Their production, design strategy and impact on the clinical trial landscape were also addressed. In the second part, the concept of multispecific antibody formats, notably MuTICEMs (Multispecific Targeted Immune Cell Engager & Modulator), as a possible answer to current immunotherapy limitations is investigated. We believe it could be the next step to take for the cancer immunotherapy research and expose why bioconjugation chemistry might play a key role in these future developments.
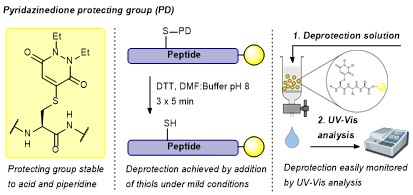
Spears, R., McMahon, C., Shamsabadi, M., Bahou, C., Thanasi, I., Rochet, L., Forte, N., Thoreau, F., Baker, J. R., Chudasama, V.*
Chem Commun, 2022, 58, 645-648.
AbstractHerein we report a thiol-labile cysteine protecting group based on an unsaturated pyridazinedione (PD) scaffold. We establish compatibility of the PD in conventional solid phase peptide synthesis (SPPS), showcasing this in the on-resin synthesis of biologically relevant oxytocin. Furthermore, we establish the applicability of the PD protecting group towards both microwave-assisted SPPS and native chemical ligation (NCL) in a model system.
Spears, R., McMahon, C., Chudasama, V.*
Chem Soc Rev, 2021, 50, 11098-11155.
AbstractProtecting group chemistry for the cysteine thiol group has enabled a vast array of peptide and protein chemistry over the last several decades. Increasingly sophisticated strategies for the protection, and subsequent deprotection, of cysteine have been developed, facilitating synthesis of complex disulfide-rich peptides, semisynthesis of proteins, and peptide/protein labelling in vitro and in vivo. In this review, we analyse and discuss the 60+ individual protecting groups reported for cysteine, highlighting their applications in peptide synthesis and protein science.
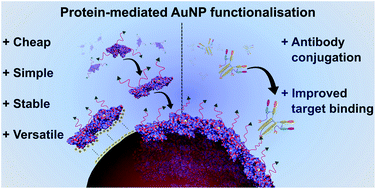
Richards, D. A., Thomas, M. R., Szijj, P. A., Foote, J., Chen, Y., Nogueira, Chudasama, V., Stevens, M. M.
Nanoscale, 2021, 13, 11921-11931.
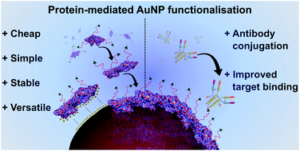
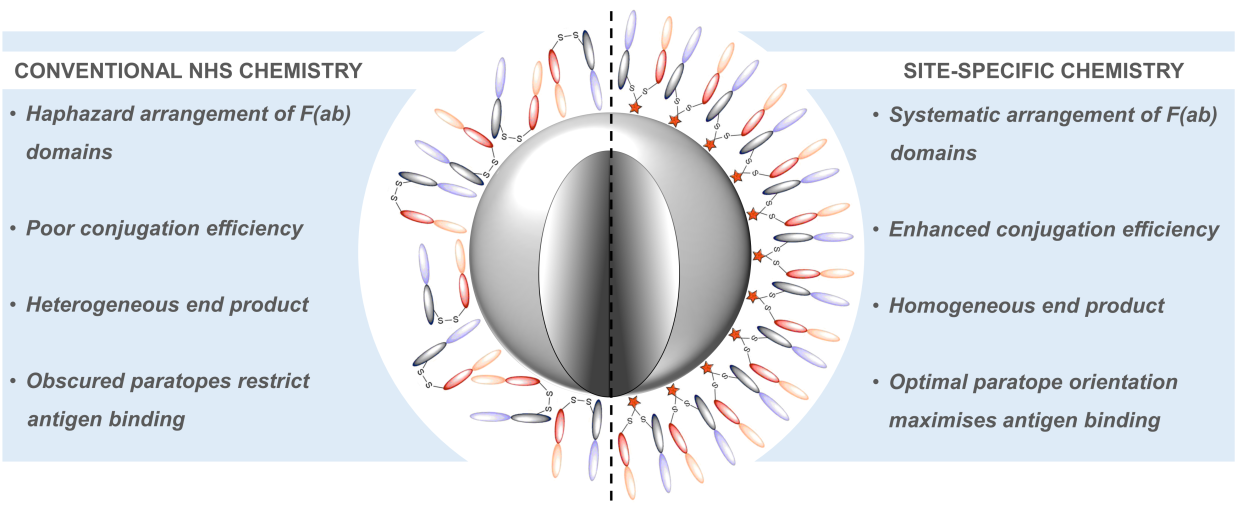
AbstractNovel methods for introducing chemical and biological functionality to the surface of gold nanoparticles serve to increase the utility of this class of nanomaterials across a range of applications. To date, methods for functionalising gold surfaces have relied upon uncontrollable non-specific adsorption, bespoke chemical linkers, or non-generalisable protein–protein interactions. Herein we report a versatile method for introducing functionality to gold nanoparticles by exploiting the strong interaction between chemically functionalised bovine serum albumin (f-BSA) and citrate-capped gold nanoparticles (AuNPs). We establish the generalisability of the method by introducing a variety of functionalities to gold nanoparticles using cheap, commercially available chemical linkers. The utility of this approach is further demonstrated through the conjugation of the monoclonal antibody Ontruzant to f-BSA–AuNPs using inverse electron-demand Diels–Alder (iEDDA) click chemistry, a hitherto unexplored chemistry for AuNP–IgG conjugation. Finally, we show that the AuNP–Ontruzant particles generated via f-BSA–AuNPs have a greater affinity for their target in a lateral flow format when compared to conventional physisorption, highlighting the potential of this technology for producing sensitive diagnostic tests.
Javaid, F., Pilotti, C., Camilli, C., Kallenberg, D., Bahou, C., Blackburn, J., Baker, J. R., Greenwood, J., Moss, S. E., Chudasama, V.*
RSC Chem Bio, 2021, 2, 1206-1220.
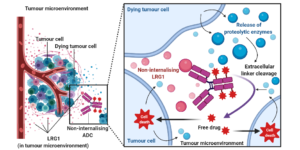
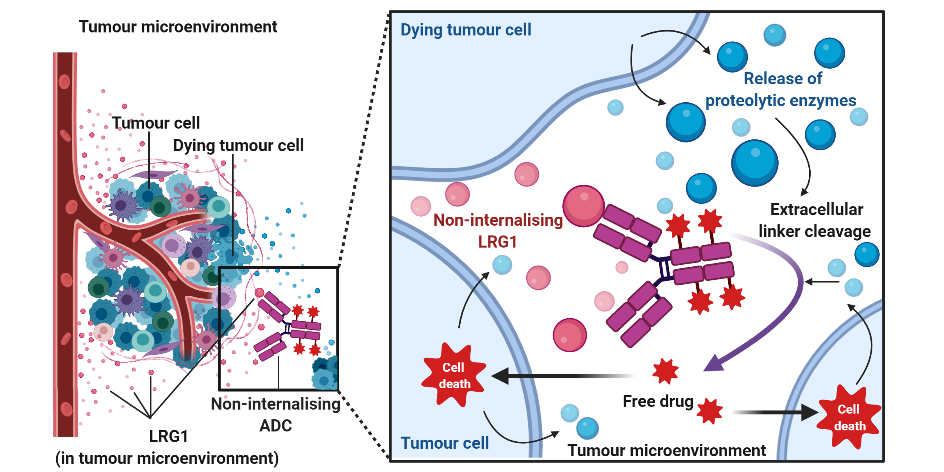
AbstractLeucine-rich alpha-2-glycoprotein 1 (LRG1) is present abundantly in the microenvironment of many tumours where it contributes to vascular dysfunction, which impedes the delivery of therapeutics. In this work we demonstrate that LRG1 is predominantly a non-internalising protein. We report the development of a novel antibody–drug conjugate (ADC) comprising the anti-LRG1 hinge-stabilised IgG4 monoclonal antibody Magacizumab coupled to the anti-mitotic payload monomethyl auristatin E (MMAE) via a cleavable dipeptide linker using the site-selective disulfide rebridging dibromopyridazinedione (diBrPD) scaffold. It is demonstrated that this ADC retains binding post-modification, is stable in serum and effective in in vitro cell studies. We show that the extracellular LRG1-targeting ADC provides an increase in survival in vivo when compared against antibody alone and similar anti-tumour activity when compared against standard chemotherapy, but without undesired side-effects. LRG1 targeting through this ADC presents a novel and effective proof-of-concept en route to improving the efficacy of cancer therapeutics.
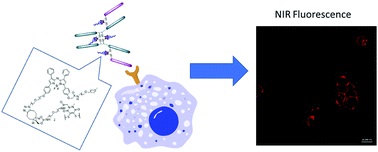
Sadraeian, M., Ferreira da Cruz, E. Boyle, R. W., Bahou, B., Chudasama, V., Janini, L. M. R., Diaz, R. S., Guimarães, F. E. G.
ACS Omega, 2021, 6, 25, 16524–16534.
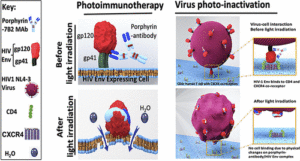
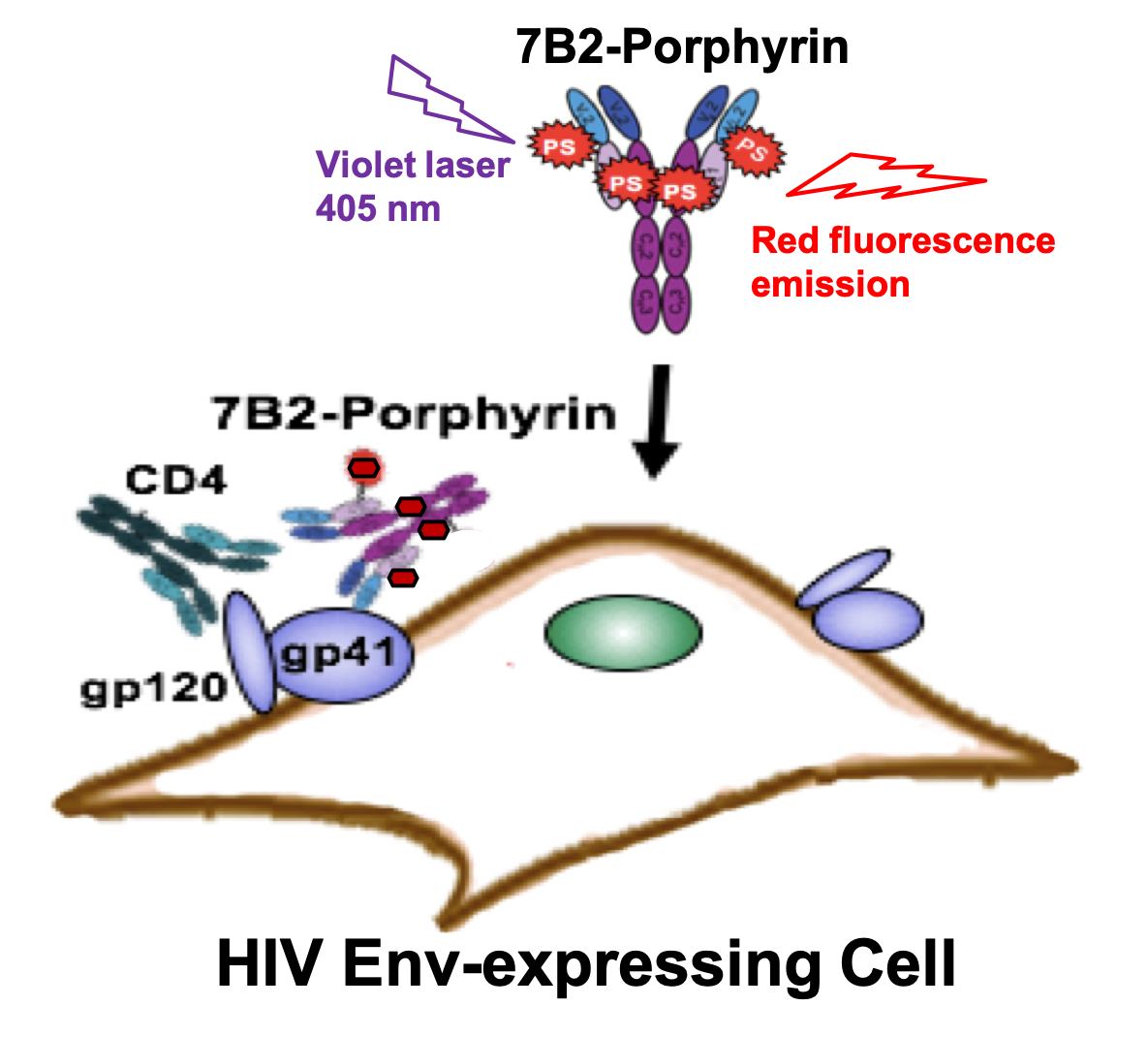
AbstractHIV-infected cells persist for decades in patients administered with antiretroviral therapy (ART). Meanwhile, an alarming surge in drug-resistant HIV viruses has been occurring. Addressing these issues, we propose the application of photoimmunotherapy (PIT) against not only HIV Env-expressing cells but also HIV. Previously, we showed that a human anti-gp41 antibody (7B2) conjugated to cationic or anionic photosensitizers (PSs) could specifically target and kill the HIV Env-expressing cells. Here, our photolysis studies revealed that the binding of photoimmunoconjugates (PICs) on the membrane of HIV Env-expressing cells is sufficient to induce necrotic cell death due to physical damage to the membrane by singlet oxygen, which is independent of the type of PSs. This finding persuaded us to study the virus photoinactivation of PICs using two HIV-1 strains, X4 HIV-1 NL4-3 and JR-CSF virus. We observed that the PICs could destroy the viral strains, probably via physical damage on the HIV envelope. In conclusion, we report the application of PIT as a possible dual-tool for HIV immunotherapy and ART by killing HIV-expressing cells and cell-free HIV, respectively.
Bahou, C., Szijj, P. A., Spears, R. J., Wall, A., Javaid, F., Sttikar, A. Love, E. A., Baker, J. R., Chudasama, V.*
Bioconjugate Chem, 2021, 32, 672-679.
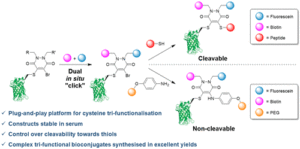
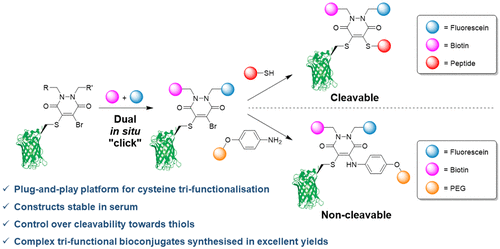
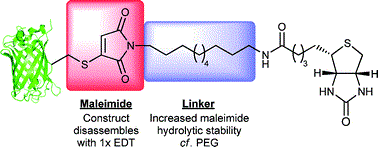
AbstractLinkers that enable the site-selective synthesis of chemically modified proteins are of great interest to the field of chemical biology. Homogenous bioconjugates often show advantageous pharmacokinetic profiles and consequently increased efficacy in vivo. Cysteine residues have been exploited as a route to site-selectively modify proteins, and many successfully approved therapeutics make use of cysteine directed conjugation reagents. However, commonly used linkers, including maleimide–thiol conjugates, are not stable to the low concentrations of thiol present in blood. Furthermore, only a few cysteine-targeting reagents enable the site-selective attachment of multiple functionalities: a useful tool in the fields of theranostics and therapeutic blood half-life extension. Herein, we demonstrate the application of the pyridazinedione motif to enable site-selective attachment of three functionalities to a protein bearing a single cysteine residue. Extending upon previously documented dual modification work, here we demonstrate that by exploiting a bromide leaving group as an additional reactive point on the pyridazinedione scaffold, a thiol or aniline derivative can be added to a protein, post-conjugation. Thiol cleavability appraisal of the resultant C–S and C–N linked thio-bioconjugates demonstrated C–S functionalized linkers to be cleavable and C–N functionalized linkers to be noncleavable when incubated in an excess of glutathione. The plug-and-play trifunctional platform was exemplified by attaching clinically relevant motifs: biotin, fluorescein, a polyethylene glycol chain, and a model peptide. This platform provides a rare opportunity to combine up to three functionalities on a protein in a site-selective fashion. Furthermore, by selecting the use of a thiol or an amine for functionalization, we provide unique control over linker cleavability toward thiols, allowing this novel linker to be applied in a range of physiological environments.
Farleigh, M.,Pham, T. T., Yu, Z., Kim, J., Sunassee, K., Firth, G., Forte, N., Chudasama, V., Baker, J. R., Long, N. J., Rivas, C., Ma, M. T.
Bioconjugate Chem, 2021, 2021, 32, 1214–1222.
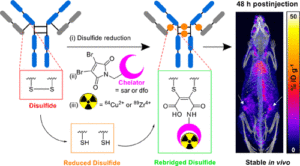
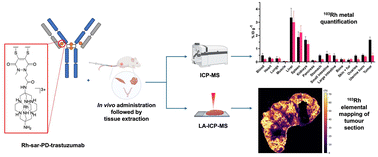
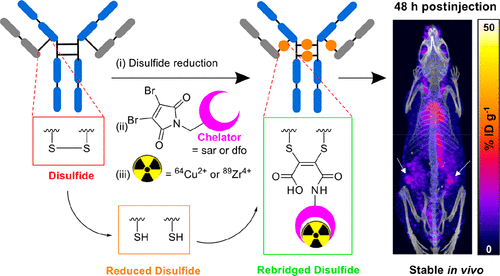
AbstractPositron Emission Tomography (PET) imaging with antibody-based contrast agents frequently uses the radioisotopes [64Cu]Cu2+ and [89Zr]Zr4+. The macrobicyclic chelator commonly known as sarcophagine (sar) is ideal for labeling receptor-targeted biomolecules with [64Cu]Cu2+. The siderophore chelator, desferrioxamine-B (dfo), has been widely used to incorporate [89Zr]Zr4+ into antibodies. Here, we describe new bifunctional chelators of sar and dfo: these chelators have been functionalized with dibromomaleimides (dbm), that enable site-specific and highly stable attachment of molecular cargoes to reduced, solvent-accessible, interstrand native disulfide groups. The new sar–dbm and dfo–dbm derivatives can be easily conjugated with the IgG antibody trastuzumab via reaction with reduced interstrand disulfide groups to give site-specifically modified dithiomaleamic acid (dtm) conjugates, sar–dtm–trastuzumab and dfo–dtm–trastuzumab, in which interstrand disulfides are rebridged covalently with a small molecule linker. Both sar– and dfo–dtm–trastuzumab conjugates have been radiolabeled with [64Cu]Cu2+ and [89Zr]Zr4+, respectively, in near quantitative radiochemical yield (>99%). Serum stability studies, in vivo PET imaging, and biodistribution analyses using these radiolabeled immunoconjugates demonstrate that both [64Cu]Cu-sar–dtm–trastuzumab and [89Zr]Zr-dfo–dtm–trastuzumab possess high stability in biological milieu. Dibromomaleimide technology can be easily applied to enable stable, site-specific attachment of radiolabeled chelators, such as sar and dfo, to native interstrand disulfide regions of antibodies, enabling tracking of antibodies with PET imaging.
Szijj, P., Chudasama, V.*
Nat Rev Chem, 2021, 5, 78-92.
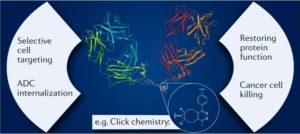
AbstractBispecific antibodies (bsAbs) target two different epitopes. These are an up-and-coming class of biologics, with two such therapeutics (emicizumab and blinatumomab) FDA approved and on the market, and many more in clinical trials. While the first reported bsAbs were constructed by chemical methods, this approach has fallen out of favour with the advent of modern genetic engineering techniques and, nowadays, the vast majority of bsAbs are produced by protein engineering. However, in recent years, relying on innovations in the fields of bioconjugation and bioorthogonal click chemistry, new chemical methods have appeared that have the potential to be competitive with protein engineering techniques and, indeed, hold some advantages. These approaches offer modularity, reproducibility and batch-to-batch consistency, as well as the integration of handles, whereby additional cargo molecules can be attached easily, e.g. to generate bispecific antibody–drug conjugates. The linker between the antibodies/antibody fragments can also be easily varied, and new formats (types, defined by structural properties or by construction methodology) can be generated rapidly. These attributes offer the potential to revolutionize the field. Here, we review chemical methods for the generation of bsAbs, showing that the newest examples of these techniques are worthy competitors to the industry-standard expression-based strategies.
2020
Sadraeian, M., Bahou, C., Ferreira da Cruz, E., Janini, L. M. R., Diaz, R. S., Boyle, R. W., Chudasama, V.*, Guimarães. F. E. G.
Int J Mol Sci, 2020, 21, 9151.
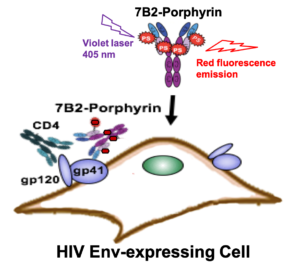
AbstractDifferent therapeutic strategies have been investigated to target and eliminate HIV-1-infected cells by using armed antibodies specific to viral proteins, with varying degrees of success. Herein, we propose a new strategy by combining photodynamic therapy (PDT) with HIV Env-targeted immunotherapy, and refer to it as HIV photoimmunotherapy (PIT). A human anti-gp41 antibody (7B2) was conjugated to two photosensitizers (PSs) with different charges through different linking strategies; “Click” conjugation by using an azide-bearing porphyrin attached via a disulfide bridge linker with a drug-to-antibody ratio (DAR) of exactly 4, and “Lysine” conjugation by using phthalocyanine IRDye 700DX dye with average DARs of 2.1, 3.0 and 4.4. These photo-immunoconjugates (PICs) were compared via biochemical and immunological characterizations regarding the dosimetry, solubility, and cell targeting. Photo-induced cytotoxicity of the PICs were compared using assays for apoptosis, reactive oxygen species (ROS), photo-cytotoxicity, and confocal microscopy. Targeted phototoxicity seems to be primarily dependent on the binding of PS-antibody to the HIV antigen on the cell membrane, whilst being independent of the PS type. This is the first report of the application of PIT for HIV immunotherapy by killing HIV Env-expressing cells.
Szijj, P. A., Kostadinova, K. A., Spears, R. J., Chudasama, V.*
Org Biomol Chem, 2020, 18, 9018-9028.
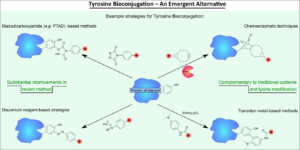
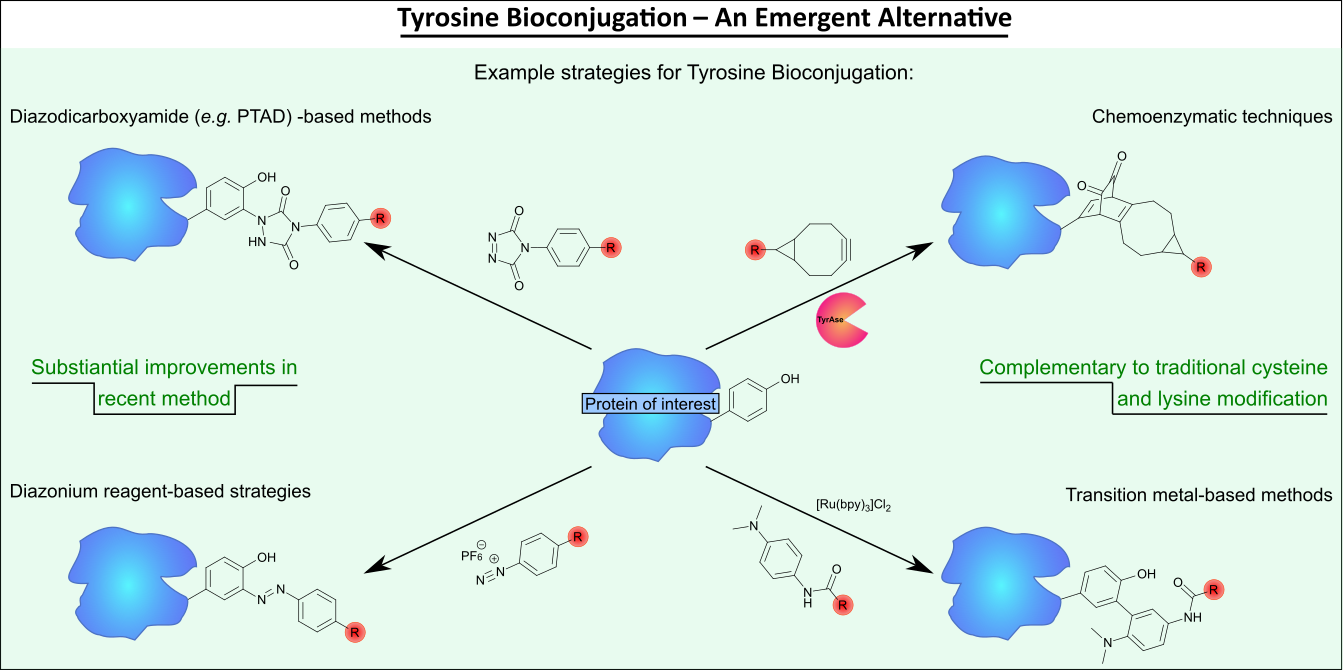
AbstractProtein bioconjugation is an increasingly important field of research, with wide-ranging applications in areas such as therapeutics and biomaterials. Traditional cysteine and lysine bioconjugation strategies are widely used and have been extensively researched, but in some cases they are not appropriate and alternatives are needed or they are not compatible with one another to enable the formation of dually (and distinctly) modified dual-conjugates (an increasingly desired class of bioconjugates). Here we review the heretofore less explored approach of tyrosine bioconjugation, which is rapidly becoming a constructive alternative/complement to the more well-established strategies. Herein we present an overview of the field, and then focus on promising recent methods that can achieve high conversion and chemoselectivity. This suggests that not only can tyrosine bioconjugation be used in conjunction with cysteine and lysine modification to obtain proteins with multiple different modifications, it is also becoming a stand-alone alternative to these more traditional methods.
Wall, A., Wills, A. G., Forte, N., Bahou, C., Bonin, L., Nicholls, K., Ma, M. T., Chudasama, V.*, Baker, J. R.
Chem Sci, 2020, 11, 11455-11460.
AbstractMaleimide chemistry is widely used in the site-selective modification of proteins. However, hydrolysis of the resultant thiosuccinimides is required to provide robust stability to the bioconjugates. Herein, we present an alternative approach that affords simultaneous stabilisation and dual functionalisation in a one pot fashion. By consecutive conjugation of a thiol and an amine to dibromomaleimides, we show that aminothiomaleimides can be generated extremely efficiently. Furthermore, the amine serves to deactivate the electrophilicity of the maleimide, precluding further reactivity and hence generating stable conjugates. We have applied this conjugation strategy to peptides and proteins to generate stabilised trifunctional conjugates. We propose that this stabilisation-dual modification strategy could have widespread use in the generation of diverse conjugates.
Greene, M. K., Chen, T., Robinson, E., Straubinger, N. L., Minx, C., Chan, D. K. W., Wang, Burrows, J. F., Van Schaeybroeck, S., Baker, J. R., Caddick, S., Longley, D. B., Mager, D. E., Straubinger, R. M. Chudasama, V.*, Scott, C. J.
Br J Cancer, 2020, 123, 1502-1512.
AbstractAntibody-drug conjugate (ADC) construction poses numerous challenges that limit clinical progress. In particular, common bioconjugation methods afford minimal control over the site of drug coupling to antibodies. Here, such difficulties are overcome through re-bridging of the inter-chain disulfides of cetuximab (CTX) with auristatin-bearing pyridazinediones, to yield a highly refined anti-epidermal growth factor receptor (EGFR) ADC.
Shamsabadi, A., Maruani, A., Ahmed, N., Chudasama, V.*
Org Biomol Chem, 2020, 18, 6258-6264.
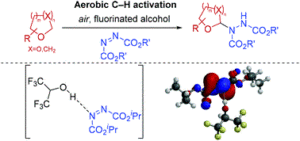
AbstractSignificant advancements in C–N bond formation via C–H bond functionalisation have made it a staple in the production of nitrogen-containing compounds in both industry and academia. However, transition metal-free synthesis, particularly in the case of C(sp3)–N formation, has remained a significant challenge to the synthetic community. Herein we report a procedure for α-C(sp3)–H amination of ethereal compounds through use of azodicarboxylates as the nitrogen source and freely-available atmospheric oxygen to access ethereal radical intermediates via aerobic C–H activation. The use of fluorinated alcohols as solvent is observed to greatly increase the efficiency of the reaction and we show experimentally and theoretically the key role of H-bonding between fluorinated alcohols and azodicarboxylates. Calculations of the condensed Fukui functions of a H-bonded fluorinated alcohol-azodicarboxylate complex correlates with a significantly increased susceptibility of azodicarboxylates to undergo reaction with radicals, which informs a number of recent reports in the literature.
Greene, M., Nogueira, J., Tracey, S. R., Richards, D. A., McDaid, W., Burrows, J. F., Campbell, K., Longley, D., Chudasama, V.*, Scott, C. J.
Nanoscale, 2020, 12, 11647-11658.
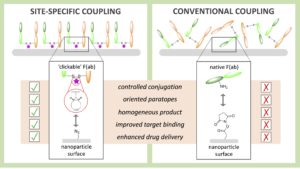
AbstractAntibody-targeted nanoparticles have shown exceptional promise as delivery vehicles for anticancer drugs, although manufacturability challenges have hampered clinical progress. These include the potential for uncontrolled and random antibody conjugation, resulting in masked or inactive paratopes and unwanted Fc domain interactions. To circumvent these issues, we show that the interchain disulfide of cetuximab F(ab) may be selectively re-bridged with a strained alkyne handle, to permit ‘click’ coupling to azide-capped nanoparticles in a highly uniform and oriented manner. When compared to conventional carbodiimide chemistry, this conjugation approach leads to the generation of nanoparticles with a higher surface loading of cetuximab F(ab) and with markedly improved ability to bind to the target epidermal growth factor receptor. Moreover, we show that entrapment of a camptothecin payload within these nanoparticles can enhance drug targeting to antigen-expressing pancreatic cancer cells, resulting in superior cytotoxicity versus the conventional nanoformulation. Collectively, this work highlights the critical need to develop refined methods for the construction of targeted nanoparticles that will accelerate their clinical translation through improved performance and manufacturability.
Petri, L., Szijj, P. A., Kelemen, A., Imre, T., Gömöry, A., Lee, M. T. W., Hegedűs, K., Ábrányi-Balogh, P., Chudasama, V.*, Kesuru, G. M.
RSC Adv, 2020, 10, 14928-14936.
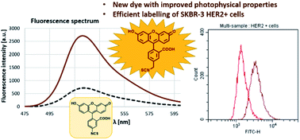
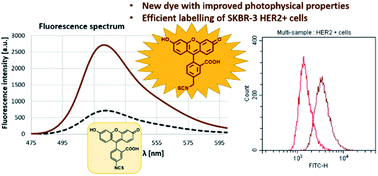
AbstractProtein labelling has a wide variety of applications in medicinal chemistry and chemical biology. In addition to covalent inhibition, specific labelling of biomolecules with fluorescent dyes is important in both target discovery, validation and diagnostics. Our research was conducted through the fragment-based development of a new benzyl-isothiocyanate-activated fluorescent dye based on the fluorescein scaffold. This molecule was evaluated against fluorescein isothiocyanate, a prevalent labelling agent. The reactivity and selectivity of phenyl- and benzyl isothiocyanate were compared at different pHs, and their activity was tested on several protein targets. Finally, the clinically approved antibody trastuzumab (and it’s Fab fragment) were specifically labelled through reaction with free cysteines reductively liberated from their interchain disulfide bonds. The newly developed benzyl-fluorescein isothiocyanate and its optimized labelling protocol stands to be a valuable addition to the tool kit of chemical biology.
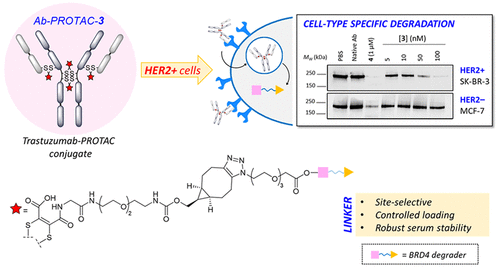
Maneiro, M., Forte, N., Shchepinova, M. M., Kounde, C. S., Chudasama, V., Baker, J. R., Tate, E. W.
ACS Chem Biol, 2020, 15, 1306-1312.
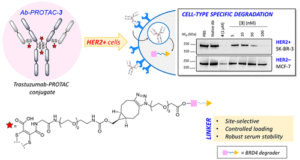
AbstractTargeting protein degradation with Proteolysis-Targeting Chimeras (PROTACs) is an area of great current interest in drug discovery. Nevertheless, although the high effectiveness of PROTACs against a wide variety of targets has been established, most degraders reported to date display limited intrinsic tissue selectivity and do not discriminate between cells of different types. Here, we describe a strategy for selective protein degradation in a specific cell type. We report the design and synthesis of a trastuzumab-PROTAC conjugate (Ab-PROTAC 3) in which E3 ligase-directed degrader activity is caged with an antibody linker which can be hydrolyzed following antibody–PROTAC internalization, releasing the active PROTAC and inducing catalytic protein degradation. We show that 3 selectively targets bromodomain-containing protein 4 (BRD4) for degradation only in HER2 positive breast cancer cell lines, while sparing HER2 negative cells. Using live cell confocal microscopy, we show internalization and lysosomal trafficking of the conjugate specifically in HER2 positive cells, leading to the release of active PROTAC in quantities sufficient to induce potent BRD4 degradation. These studies demonstrate proof-of-concept for tissue-specific BRD4 degradation, overcoming limitations of PROTAC selectivity, with significant potential for application to novel targets.
Maruani, A., Szijj, P. A., Bahou, C., Nogueira, J. C. F., Caddick, S., Baker, J. R., Chudasama, V.*
Bioconjugate Chem, 2020, 31, 520-529.
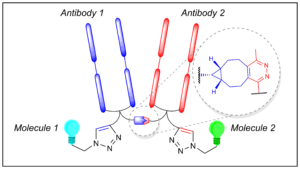
AbstractDiseases are multifactorial, with redundancies and synergies between various pathways. However, most of the antibody-based therapeutics on the market interact with only one target, thus limiting their efficacy. The targeting of multiple epitopes could improve the therapeutic index of treatment and counteract mechanisms of resistance. To this effect, a new class of therapeutics has emerged: bispecific antibodies. Bispecific formation using chemical methods is rare and low-yielding and/or requires a large excess of one of the two proteins to avoid homodimerization and heterogeneity. In order for chemically prepared bispecifics to deliver their full potential, high-yielding, modular, and reliable cross-linking technologies are required. Herein, we describe a novel approach not only for the rapid and high-yielding chemical generation of bispecific antibodies from native antibody fragments, but also for the site-specific dual functionalization of the resulting bioconjugates. Based on orthogonal clickable functional groups, this strategy enables the assembly of functionalized bispecifics with controlled loading in a modular and convergent manner.
Xu, L., Raabe, M.., Zegota, M. M., Nogueira, J. C. F., Chudasama, V., Kuan, S. L., Weil, T.
Org Biomol Chem, 2020, 18, 1140-1147.
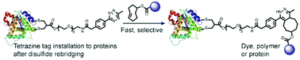
AbstractAn inverse electron demand Diels–Alder reaction between tetrazine and trans-cyclooctene (TCO) holds great promise for protein modification and manipulation. Herein, we report the design and synthesis of a tetrazine-based disulfide rebridging reagent, which allows the site-selective installation of a tetrazine group into disulfide-containing peptides and proteins such as the hormone somatostatin (SST) and the antigen binding fragment (Fab) of human immunoglobulin G (IgG). The fast and efficient conjugation of the tetrazine modified proteins with three different TCO-containing substrates to form a set of bioconjugates in a site-selective manner was successfully demonstrated for the first time. Homogeneous, well-defined bioconjugates were obtained underlining the great potential of our method for fast bioconjugation in emerging protein therapeutics. The formed bioconjugates were stable against glutathione and in serum, and they maintained their secondary structure. With this work, we broaden the scope of tetrazine chemistry for site-selective protein modification to prepare well-defined SST and Fab conjugates with preserved structures and good stability under biologically relevant conditions.
Nogueira, J. C. F., Paliashvili, K., Bradford, A., Di Maggio, F., Richards, D. A, Day, R., Chudasama, V.*
Org Biomol Chem, 2020, 18, 2215-2218.
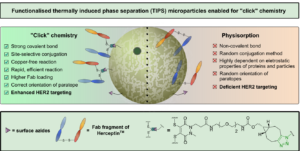
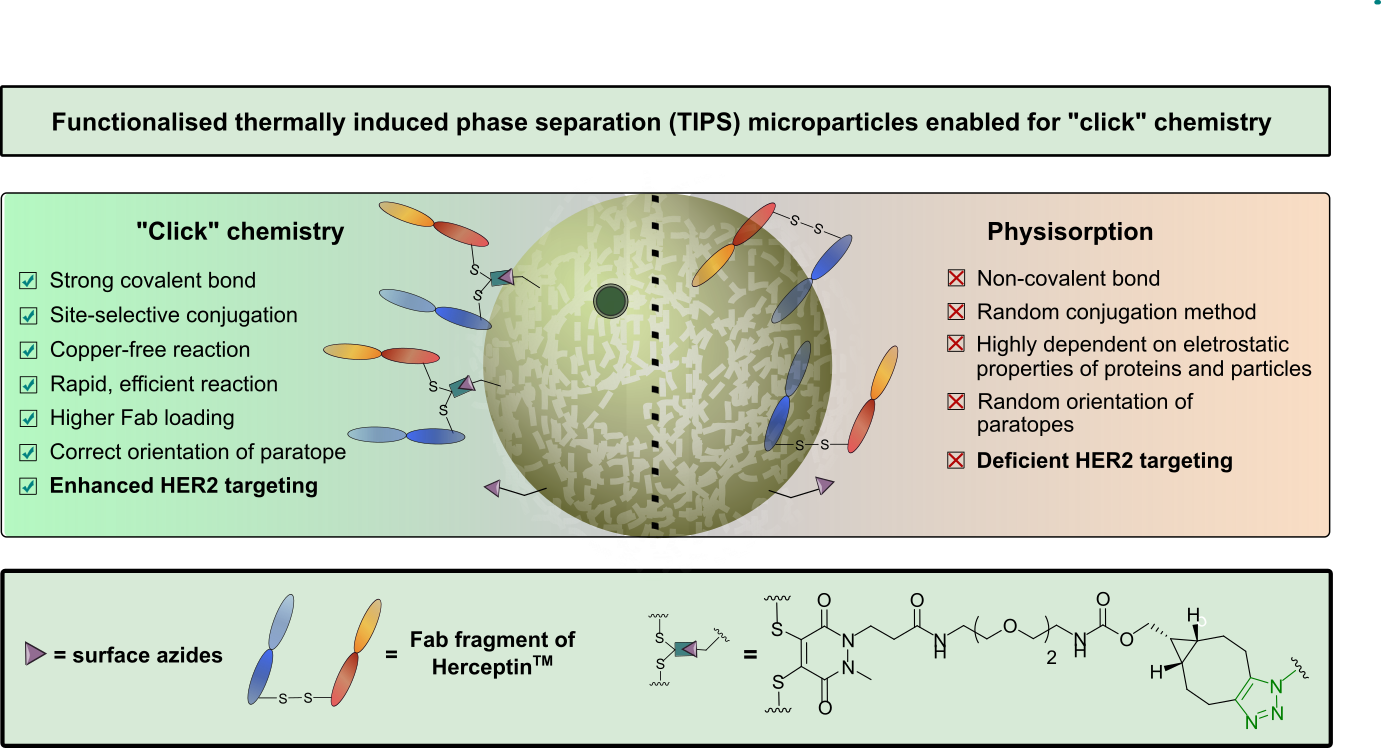
AbstractDue to their homogeneity, tuneable properties, low cost and ease of manufacture, thermally induced phase separation (TIPS) polymeric microparticles are emerging as an exciting class of injectable device for the treatment of damaged tissue or complex diseases, such as cancer. However, relatively little work has explored enhancing surface functionalisation of this system. Herein, we present the functionalisation of TIPS microparticles with both small molecules and an antibody fragment of Herceptin™, via a heterobifunctional pyridazinedione linker capable of participating in SPAAC “click” chemistry, and compare it to the traditional method of preparing active-targeted microparticle systems, that is, physisorption of antibodies to the microparticle surface. Antigen-binding assays demonstrated that functionalisation of microparticles with Herceptin Fab, via a pyridazinedione linker, provided an enhanced avidity to HER2+ when compared to traditional physisorption methods.
72. Fine-tuning thio-pyridazinediones as SMDC scaffolds (with intracellular thiol release via a novel self-immolative linker)
Fernandez, M., Shamsabadi, A., Chudasama, V.*
Chem Commun, 2020, 56, 1125-1128.
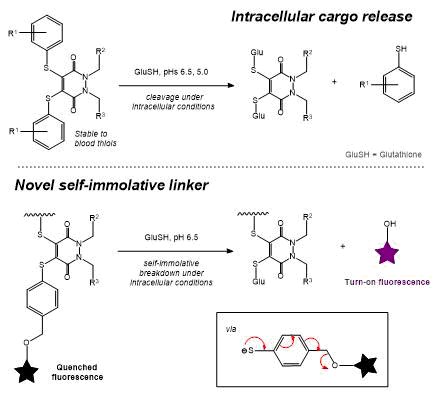
AbstractHerein we report the synthesis of a library of thioalkyl- and thioaryl-pyridazinediones for thiol-based self-immolative release of cargo. A bisthioaryl-pyridazinedione is shown to be stable to serum protein albumin but unstable in intracellular conditions. A derivatised analogue underwent self-immolative degradation in cellular thiol conditions as evidenced by LC-MS/release of a turn-on fluorescence fluorophore; versatility of the thiol-pyridazinedione is demonstrated through synthesis of SMDC precursors that contain three different functional groups on the same central molecule.
2019
Ahmed, N., Shamsabadi, A., Chudasama, V.*
ACS Omega, 2019, 4, 22601-22612.
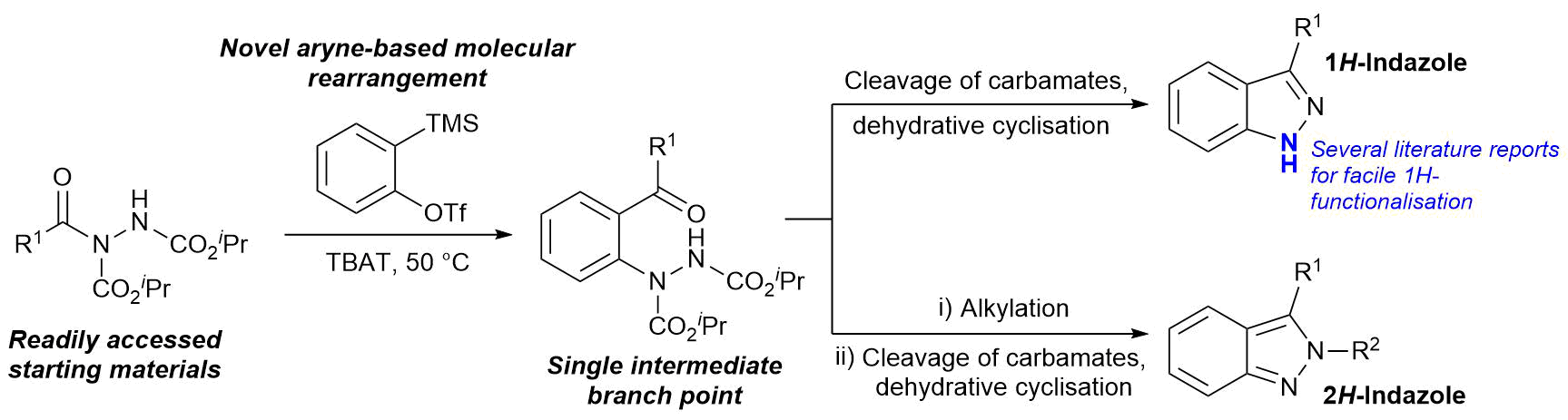
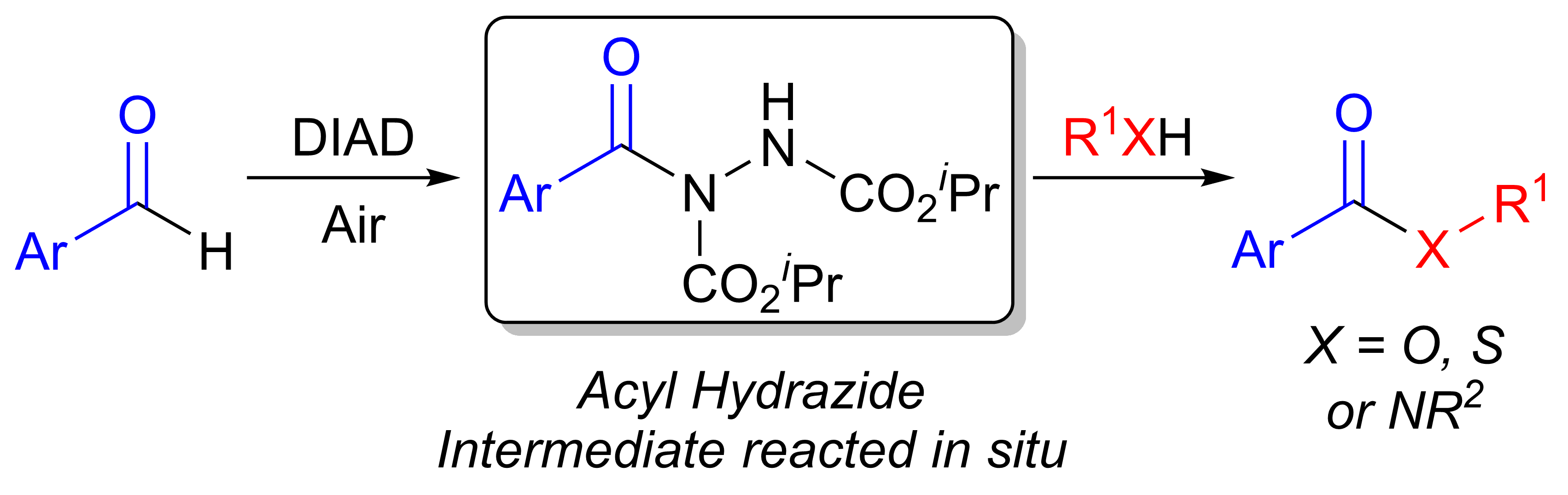
AbstractHerein, we report the transformation of readily accessed acyl hydrazides into protected 2-aminobenzophenones via a two-step process involving an aryne-based molecular rearrangement followed by a one-pot addition–elimination procedure. The assembly of the scaffold is tolerant of a wide variety of functional groups, and the carbamate group on the product can be facilely removed to afford highly valuable 2-aminobenzophenones. Application of the protocol was demonstrated in the synthesis of neurological medicine phenazepam.
70. Use of pyridazinediones as extracellular cleavable linkers through reversible cysteine conjugation
Bahou, C.,^ Spears, R. J.,^ Aliev, A. E., Maruani, A., Fernandez, M., Javaid, F., Szijj, P. A., Baker, J. R., Chudasama, V.* (^ joint 1st authors)
Chem Commun, 2019, 55, 14829-14832.
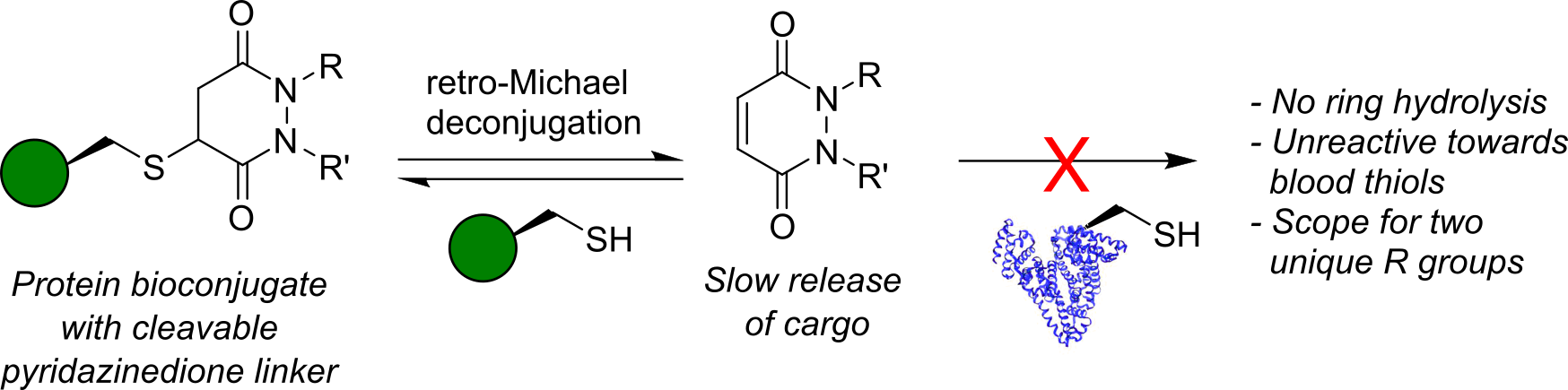
AbstractHerein we report a retro-Michael deconjugation pathway of thiol–pyridazinedione linked protein bioconjugates to provide a novel cleavable linker technology. We demonstrate that the novel pyridazinedione linker does not suffer from off-target modification with blood thiols (e.g., glutathione, human serum albumin (HSA)), which is in sharp contrast to an analogous maleimide linker.
69. Controlling Engineered P2X Receptors with Light
Atkinson, B. N., Chudasama, V., Browne, L. E.
Purinergic Signaling, 2019, pp 301-309.
AbstractThis chapter details methods to express and modify ATP-gated P2X receptor channels so that they can be controlled using light. Following expression in cells, a photoswitchable tool compound can be used to covalently modify mutant P2X receptors, as previously demonstrated for homomeric P2X2 and P2X3 receptors, and heteromeric P2X2/3 receptors. Engineered P2X receptors can be rapidly and reversibly opened and closed by different wavelengths of light. Light-activated P2X receptors can be mutated further to impart ATP-insensitivity if required. This method offers control of specific P2X receptor channels with high spatiotemporal precision to study their roles in physiology and pathophysiology.
68. Cysteine-To-Lysine Transfer Antibody Fragment Conjugation
Forte, N., Benni, I., Karu, K., Chudasama, V.*, Baker, J. R.
Chem Sci, 2019, 10, 10919-10924.
AbstractThe modification of lysine residues with acylating agents has represented a ubiquitous approach to the construction of antibody conjugates, with the resulting amide bonds being robustly stable and clinically validated. However, the conjugates are highly heterogeneous, due to the presence of numerous lysines on the surface of the protein, and greater control of the sites of conjugation are keenly sought. Here we present a novel approach to achieve the targeted modification of lysines distal to an antibody fragment’s binding site, using a disulfide bond as a temporary ‘hook’ to deliver the acylating agent. This cysteine-to-lysine transfer (CLT) methodology offers greatly improved homogeneity of lysine conjugates, whilst retaining the advantages offered by the formation of amide linkages.
67. Optimised approach to albumin–drug conjugates using monobromomaleimide-C-2 linkers
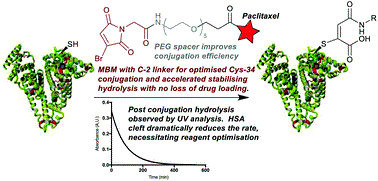
Wall, A., Nicholls, K., Caspersen, M.,Skrivergaard, S., Howard, K.A., Karu, K., Chudasama, V.*, Baker, J.R.
Org Biomol Chem, 2019, 17, 7870-7873.
AbstractConjugation of therapeutics to human serum albumin (HSA) using bromomaleimides represents a promising platform for half-life extension. We show here that the Cys-34 crevice substantially reduces the rate of serum stabilising maleimide hydrolysis in these conjugates, necessitating reagent optimisation. This improved reagent design is applied to the construction of an HSA-paclitaxel conjugate, preventing drug loss during maleimide hydrolysis.
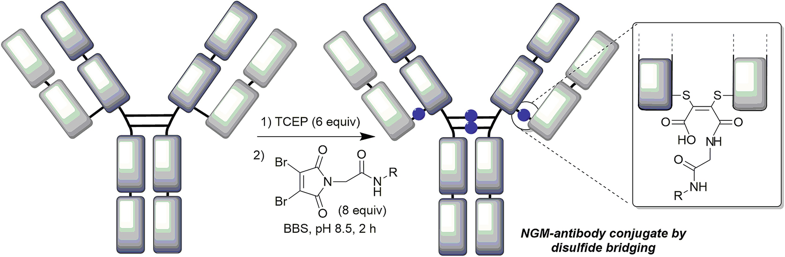
66. Application of Next-Generation Maleimides (NGMs) to Site-Selective Antibody Conjugation
Forte, N., Morais, M., Chudasama, V., Baker, J.R.
Bioconjugation. Methods in Molecular Biology, vol 2033. Humana, New York, NY, pp 15-24.
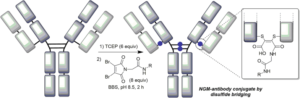
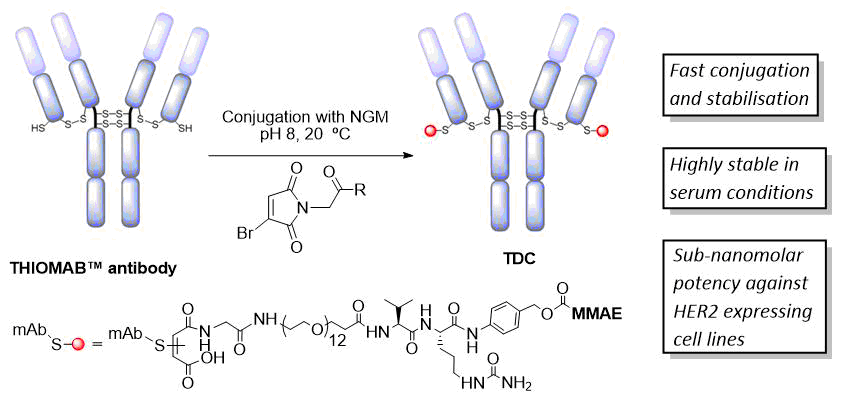
AbstractSite-selective antibody conjugation is widely recognized as a key strategy for the optimum construction of antibody–drug conjugates (ADCs). Achieving such bioconjugation directly onto native antibodies would represent the ideal solution, as it would afford greatly improved homogeneity whilst avoiding the need for genetic engineering, and even allow the repurposing of existing antibodies “off-the shelf.” Here we describe a protocol for the use of next-generation maleimides (NGMs) for the selective modification of the four interchain disulfide bonds present in a typical IgG1 antibody format. These reagents retain the efficiency of classical maleimides whilst serving to rebridge each reduced disulfide bond, affording one attachment per disulfide. The approach is simple, uses readily available reagents, and generates robustly stable conjugates which are ideal for in vitro or in vivo applications. In addition to use in the construction of ADCs these reagents can also be used to develop antibody conjugates for imaging, bispecifics, and broadly for use across biology and medicine.
65. Oriented attachment of VNAR proteins, via site-selective modification, on PLGA-PEG nanoparticles enhances nanoconjugate performance
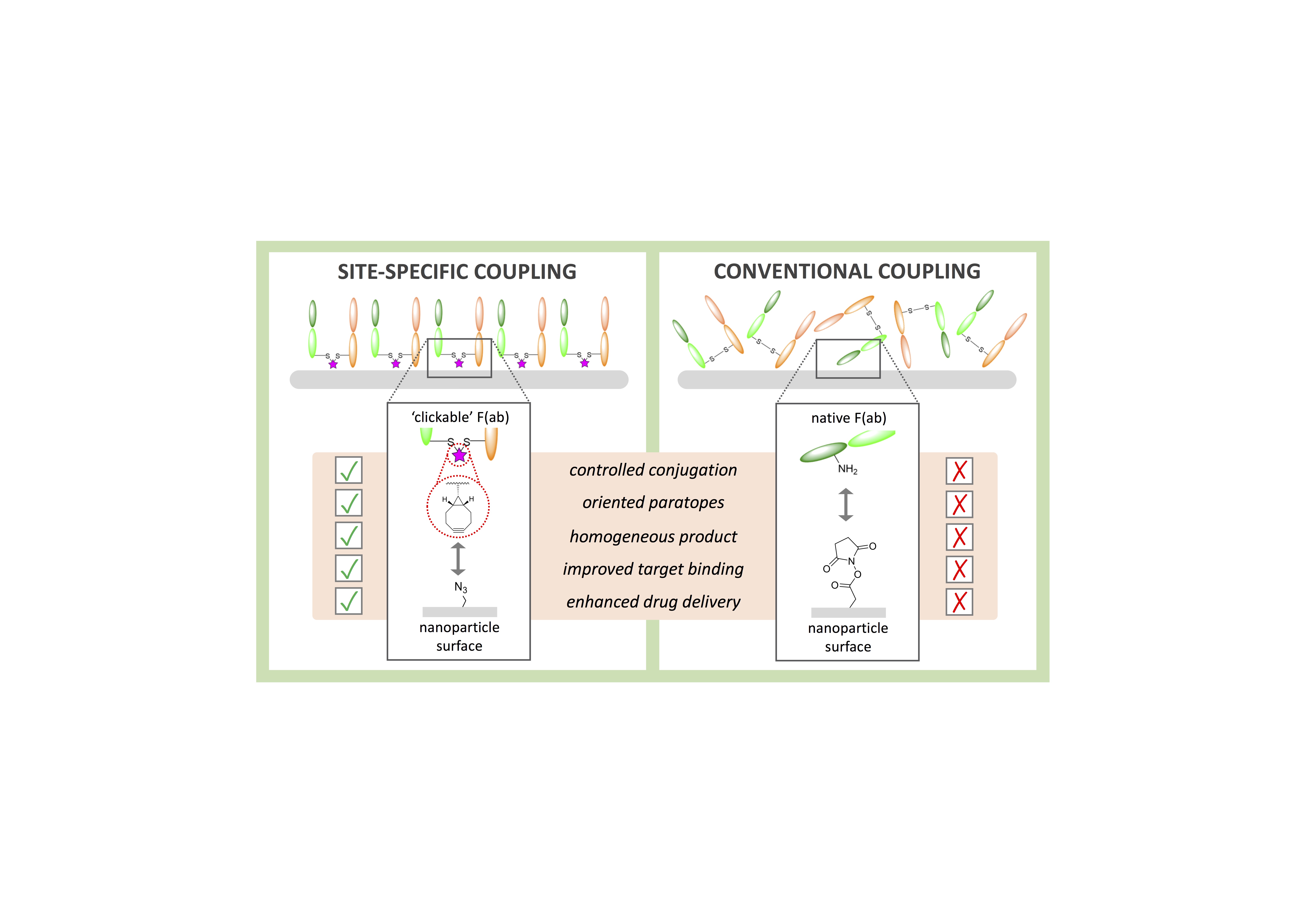
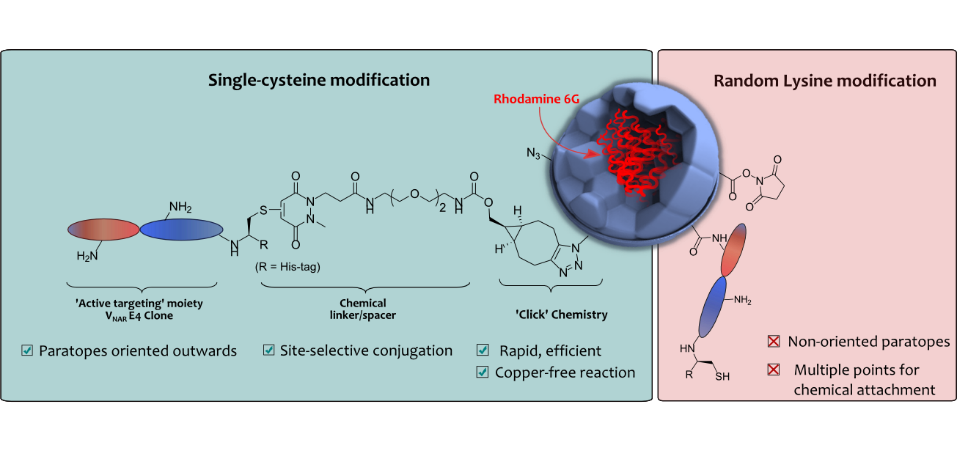
Nogueira, J., Greene, M., Richards, D. A., Furby, A. O., Steven, J., Porter, A., Barelle, C., Scott, C. J., Chudasama, V.*
Chem Commun, 2019, 55, 7671-7674.
AbstractHerein we report the construction of a nanoparticle-based drug delivery system which targets a key regulator in tumour angiogenesis. We exploit a Variable New Antigen Receptor (VNAR) domain, conjugated using site-specific chemistry, to direct PLGA-PEG nanoparticles to DLL4. The importance of site-specific chemistry for achieving high avidity in these systems is demonstrated.
64. Disulfide Modified IgG1: An Investigation of Biophysical Profile and Clinically Relevant Fc Interactions
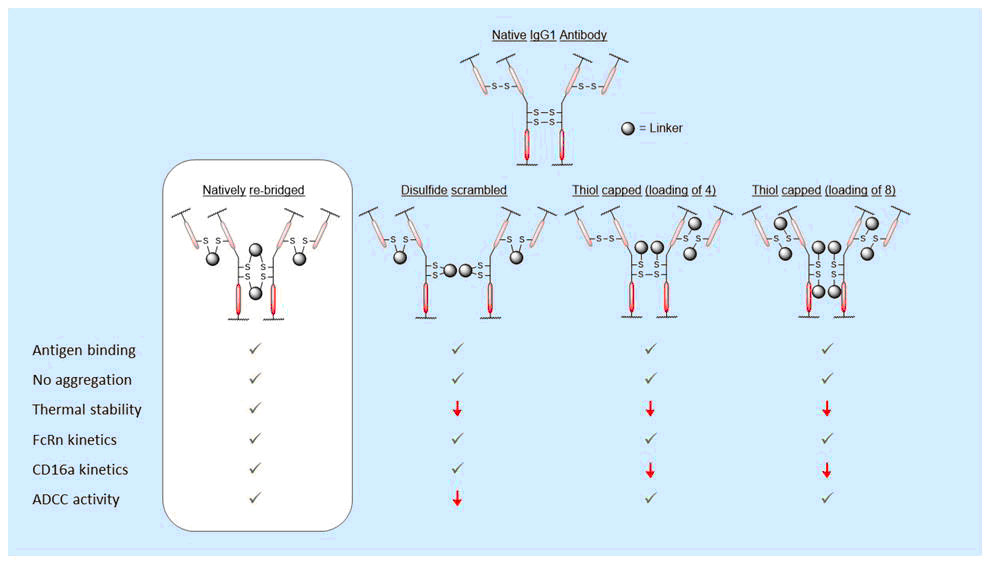
Bahou, C., Love, E., Leonard, S., Spears, R. J., Maruani, A., Armour, K., Baker, J. R., Chudasama, V.*
Bioconjugate Chem, 2019, 30, 1048-1054.
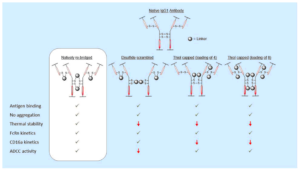
AbstractModification of immunoglobulin G (IgG) 1 proteins in cancer treatment is a rapidly growing field of research. Antibody–drug conjugates (ADCs) exploit the targeted nature of this immunotherapy by conjugating highly potent drugs to antibodies, allowing for effective transport of cargo(s) to cancerous cells. Of the many bioconjugation strategies now available for the formation of highly homogenous ADCs, disulfide modification is considered an effective, low-cost and widely accepted method for modifying IgG1s for improved clinical benefit. However, little is known about how disulfide modification impacts clinically relevant fragment crystallisable (Fc) region interactions. Although often overlooked as a secondary ADC function, Fc interactions could prove key in rational design of cancer cell-targeting ADCs through consideration of potent mechanisms such as antibody-dependant cellular cytotoxicity (ADCC). This work explores different IgG1 disulfide modification techniques and the effect they have on quantifiable secondary IgG1 Fc interactions (e.g. CD16a and FcRn). The solvent accessible disulfide residues of trastuzumab, a clinically relevant IgG1, were modified to provide a range of bioconjugates with differing amounts of inter-chain covalent linkages. It was found that by natively re-bridging the IgG1 model, all tested Fc functionalities were not significantly affected. Additionally, in non Fc-specific biophysical experiments (e.g. thermal stability/aggregation), the natively re-bridged species provided an exceptional profile, showing no significant change from the tested native antibody. Conjugates with significant disruption of the covalent connectivity of IgG1 chains resulted in a suboptimal Fc profile (CD16a kinetics or ADCC activity), in addition to sub-standard non Fc-specific attributes (thermal stability). These results advocate native disulfide re-bridging as an excellent synthetic strategy for forming homogenous IgG1 bioconjugates, with no reported negative impact on biophysical profile relative to the native antibody.
Shamsabadi, A., Chudasama, V.*
Org Biomol Chem, 2019, 17, 2865-2872.
AbstractHerein we describe recent developments in selective, metal-free, dioxygen-induced C–H activation. This method of activating C–H bonds is an attractive alternative to traditional methodologies as it uses dioxygen, an inherently sustainable and widely accessible oxidant, in place of expensive or toxic metals and/or hazardous peroxides. Reactions developed on the basis of using aerobic C–H activation are also discussed.
2018
Galan, S. R. G., Wickens, J. R., Dadova, J., Ng, W.-L., Zhang, X., Simion, R. A., Quinlan, R., Pires, E., Paton, R. S., Caddick, S., Chudasama, V.*, Davis, B. G.
Nature Chem Bio, 2018, 14, 955-963.
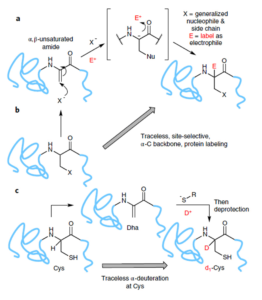
AbstractIsotopic replacement has long-proven applications in small molecules. However, applications in proteins are largely limited to biosynthetic strategies or exchangeable (for example, N–H/D) labile sites only. The development of postbiosynthetic, C–1H → C–2H/D replacement in proteins could enable probing of mechanisms, among other uses. Here we describe a chemical method for selective protein α-carbon deuteration (proceeding from Cys to dehydroalanine (Dha) to deutero-Cys) allowing overall 1H→2H/D exchange at a nonexchangeable backbone site. It is used here to probe mechanisms of reactions used in protein bioconjugation. This analysis suggests, together with quantum mechanical calculations, stepwise deprotonations via on-protein carbanions and unexpected sulfonium ylides in the conversion of Cys to Dha, consistent with a ‘carba-Swern’ mechanism. The ready application on existing, intact protein constructs (without specialized culture or genetic methods) suggests this C–D labeling strategy as a possible tool in protein mechanism, structure, biotechnology and medicine.
Chudasama, V.*
Drug Discov Today Technol., 2018, 30, 1-2.
Forte, N., Chudasama, V.*, Baker J. R.
Drug Discov Today Technol., 2018, 30, 11-20.
AbstractAntibody-drug conjugates (ADCs) constructed using site-selective labelling methodologies are likely to dominate the next generation of these targeted therapeutics. To this end, disulfide bridging has emerged as a leading strategy as it allows the production of highly homogeneous ADCs without the need for antibody engineering. It consists of targeting reduced interchain disulfide bonds with reagents which reconnect the resultant pairs of cysteine residues, whilst simultaneously attaching drugs. The 3 main reagent classes which have been exemplified for the construction of ADCs by disulfide bridging will be discussed in this review; bissulfones, next generation maleimides and pyridazinediones, along with others in development.
59. A facile route to 1H– and 2H-indazoles from readily accessed acyl hydrazides by exploiting a novel aryne-based molecular rearrangement
Shamsabadi, A., Chudasama, V.*
Chem Commun, 2018, 54, 11180-11183.
AbstractHerein we report the transformation of readily synthesised/facilely accessed acyl hydrazides into 2-hydrazobenzophenones via a novel molecular rearrangement using aryne chemistry. The developed reaction protocol is performed under relatively mild conditions, is tolerant of a wide variety of functional groups, and the 2-hydrazobenzophenone products provide access to both 1H– and 2H-indazoles from a single intermediate.
58. Minireview: Addressing the retro-Michael instability of maleimide bioconjugates
Szijj, P. A., Bahou, C., Chudasama, V.*
Drug Discov. Today Technol., 2018, 30, 27-34.
AbstractBioconjugation, the modification of biological macromolecules such as proteins, is an up and coming area in the field of chemical biology. Antibody-drug conjugates (ADCs), combining the antigen-selectivity of natural antibodies with the cytotoxic potency of small molecule drugs, are a powerful therapeutic technology. Four such constructs are currently on the market for cancer therapy. However, the conjugation methodology employed in these therapeutics is far from ideal. Herein we provide an overview on methods that attempt to increase the safety and efficacy of ADCs via “self-hydrolysing maleimides” or by improving the stability of maleimide-conjugates by other means. We find that some very promising reagents have been reported, however the mechanism by which each of these reagents acts is not clear, thus limiting rational design for some strategies.
57. Tuning the Hydrolytic Stability of Next Generation Maleimide Cross-Linkers Enables Access to Albumin-Antibody Fragment Conjugates and tri-scFvs
Forte, N., Livanos, M., Miranda, E., Morais, M., Yang, X., Rajkumar, V.S., Chester, K.A.,Chudasama, V.*, Baker, J.R.
Bioconjugate Chem, 2018, 29, 486-492.
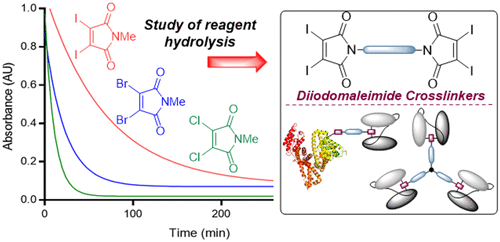
AbstractWe describe investigations to expand the scope of next generation maleimide cross-linkers for the construction of homogeneous protein–protein conjugates. Diiodomaleimides are shown to offer the ideal properties of rapid bioconjugation with reduced hydrolysis, allowing the cross-linking of even sterically hindered systems. The optimized linkers are exploited to link human serum albumin to antibody fragments (Fab or scFv) as a prospective half-life extension platform, with retention of antigen binding and robust serum stability. Finally, a triprotein conjugate is formed, by linking scFv antibody fragments targeting carcinoembryonic antigen. This tri-scFv is shown to infer a combination of greater antigen avidity and increased in vivo half-life, representing a promising platform for antibody therapeutic development.
56. Highly homogeneous antibody modification through optimisation of the synthesis and conjugation of functionalised dibromopyridazinediones
Bahou, C., Richards, D.A., Maruani, A., Love, E.A., Javaid, F., Caddick,S., Baker, J.R, Chudasama, V.*
Org Biomol Chem, 2018, 16, 1359-1366.
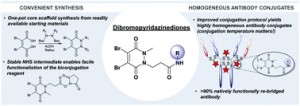
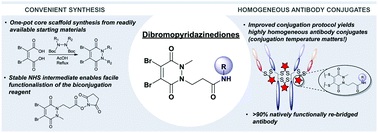
AbstractDue to their exquisite cysteine-selectivity, excellent stability, and ability to functionally rebridge disulfide bonds, dibromopyridazinediones are emerging as an exciting new class of bioconjugation reagents, particularly in the field of antibody conjugation. Despite this, relatively little work has been performed on the optimisation of their synthesis and subsequent reaction with immunoglobulins. Herein we present a novel synthetic route towards functionalised dibromopyridazinediones, proceeding via an isolatable dibromopyridazinedione-NHS ester. Reaction of this activated intermediate with a variety of amines produces functional dibromopyridazinediones in good to excellent yields. The disulfide rebridging capacity of these reagents was optimised on the clinically relevant IgG1 trastuzumab, resulting in a general method which allows for the generation of site-selectively modified native trastuzumab with over 90% homogeneity (no disulfide scrambling) without the need for protein engineering or enzymatic conjugation.
55. Synthesis of a novel HER2 targeted aza-BODIPY–antibody conjugate: Synthesis, photophysical characterisation and in vitro evaluation
Cheng, M., Maruani, A., Savoie, H., Chudasama, V.*, Boyle, R.W.
Org Biomol Chem, 2018, 16, 1144-1149.
AbstractWe herein report the synthesis and analysis of a novel aza-BODIPY–antibody conjugate, formed by controlled and regioselective bioconjugation methodology. Employing the clinically relevant antibody, which targets HER2 positive cancers, represents the first example of an antibody targeting strategy for this class of near-IR emitting fluorophore. The NIR fluorescence and binding properties were validated through in vitro studies using live cell confocal imaging.
54. Advances in Targeting the Folate Receptor in the Treatment/Imaging of Cancers
Fernandez, M., Javaid, F., Chudasama, V.*
Chem Sci, 2018, 9, 790-810.

AbstractThe folate receptor (FR) is a recognised biomarker for tumour cells due to it being overexpressed on a large number of tumours. Consequently, the FR has been exploited by many diagnostic and therapeutic tools to allow targeted delivery to, and imaging of, cancer cells. Herein, we describe the many different approaches by which this has been achieved, including the attachment of folate to potent chemotherapeutic drugs to form FR-targeting small molecule drug conjugates (SMDCs), FR-targeting antibodies (as antibody alone and as an antibody-drug conjugate), and in the form of complementary nanotechnology-folate platforms; as well as imaging variants thereof. The potential of exploiting the FR for targeted therapy/imaging has the potential to revolutionise the way several cancers are treated. These FR-targeted technologies can also pave the way for inspiring further sophisticated drug conjugates, especially as this receptor is being targeted by use of several complementary technologies: small molecule, nanoparticle and protein-based – thus providing broad and distinct knowledge in the area.
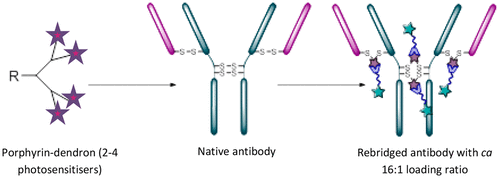
53. Assembly of high-potency photosensitiser-antibody conjugates through application of dendron multiplier technology
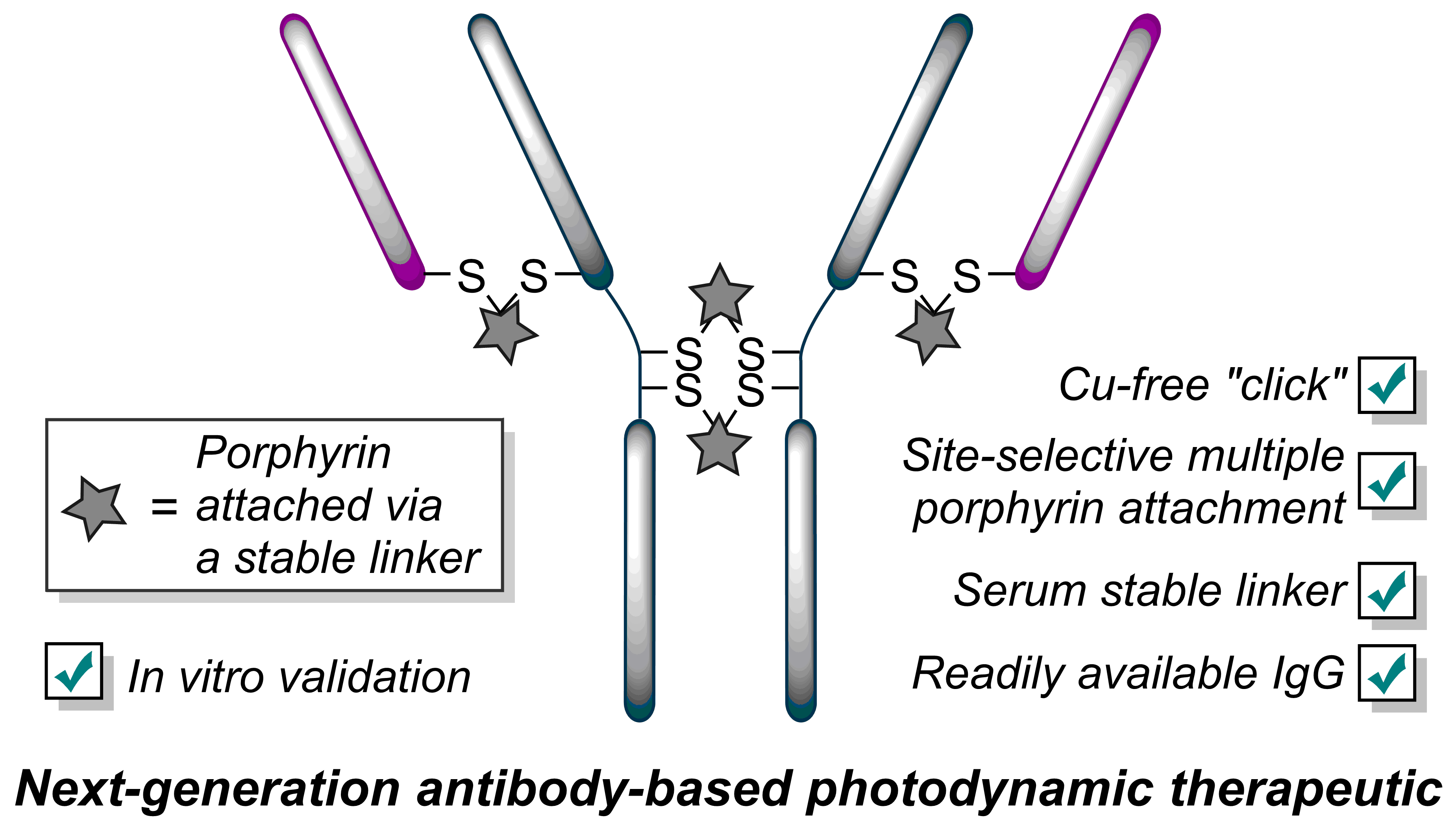
Bryden, F., Maruani, A., Rodrigues, J.M.M., Cheng, M.H.Y., Savoie, H., Beeby, A., Chudasama, V.*, Boyle, R.W.
Bioconjugate Chem, 2018, 29, 176-181.
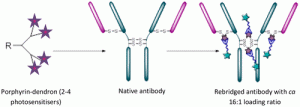
AbstractExploitation of photosensitisers as payloads for antibody-based anti-cancer therapeutics offers a novel alternative to the small pool of commonly-utilised cytotoxins. However, existing bioconjugation methodologies are incompatible with the requirement of increased antibody loading without compromising antibody function, stability or homogeneity. Herein, we describe the first application of dendritic multiplier groups to allow the loading of more than 4 porphyrins to a full IgG antibody in a site-specific and highly homogeneous manner. Photophysical evaluation of UV-visible absorbance and singlet oxygen quantum yields highlighted porphyrin-dendron 14 as the best candidate for bioconjugation; with subsequent bioconjugation producing a HER2-targeted therapeutic with average loading ratios of 15.4:1. In vitro evaluation of conjugate 18 demonstrated a nanomolar photocytotoxic effect in a target cell line, which overexpresses HER2, with no observed photocytotoxicity at the same concentration in a control cell line which expresses native HER2 levels, or in the absence of irradiation with visible light.
Loynachan, C.N., Thomas, M.R., Gray, E.R., Richards, D.A., Kim, J., Miller, B.S., Brookes, J.C., Agarwal, S., Chudasama, V., McKendry, R.A., Stevens, M.M.
ACS Nano, 2018, 12, 279-288.
AbstractPaper-based lateral flow immunoassays (LFIAs) are one of the most widely used point-of-care (PoC) devices, however, their application in early disease diagnostics is often limited due to insufficient sensitivity for the requisite sample sizes and the short timeframes of PoC testing. To address this, we developed a serum-stable, nanoparticle catalyst-labelled LFIA with a sensitivity surpassing that of both current commercial and published sensitivities for paper-based detection of p24, one of the earliest and most conserved biomarkers of HIV. We report the synthesis and characterization of porous platinum core-shell nanocatalysts (PtNCs), which show high catalytic activity when exposed to complex human blood serum samples. We explored the application of antibody functionalized PtNCs with strategically and orthogonally modified nanobodies with high affinity and specificity toward p24 and establish the key larger nanoparticle size regimes needed for efficient amplification and performance in LFIA. Harnessing the catalytic amplification of PtNCs enabled naked-eye detection of p24 spiked into sera in the low femtomolar range (ca. 0.8 pg•mL-1), and the detection of acute phase HIV in clinical human plasma samples in under 20 minutes. This provides a versatile absorbance-based and rapid LFIA with sensitivity capable of significantly reducing the HIV acute phase detection window. This diagnostic may be readily adapted for detection of other biomolecules; as an ultrasensitive screening tool for infectious and non-communicable diseases, and can be capitalized upon in PoC settings for early disease detection.
51. Generating Next-Generation Antibody-Nanoparticle Conjugates through the Oriented Installation of Non-Engineered Antibody Fragments
Greene, M., Richards, D.A., Nogueira, J., Campbell, K., Smyth, P., Fernandez, M., Scott, C.J., Chudasama, V.*
Chem Sci, 2018, 9, 79-87.
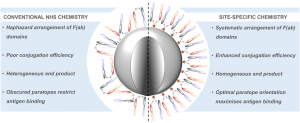
AbstractThe successful development of targeted nanotherapeutics is contingent upon the conjugation of therapeutic nanoparticles to target-specific ligands, with particular emphasis placed on antibody-based ligands. Thus, new methods which enable the covalent and precise installation of targeting antibodies to nanoparticle surfaces are greatly desired, especially those which do not rely on costly and time-consuming antibody engineering techniques. Herein we present a novel method for the highly controlled and oriented covalent conjugation of non-engineered antibody F(ab) fragments to PLGA-PEG nanoparticles using disulfide-selective pyridazinedione linkers and strain-promoted alkyne-azide click chemistry. Exemplification of this method with trastuzumab and cetuximab showed significant improvements in both conjugation efficiency and antigen binding capability, when compared to commonly employed strategies for antibody-nanoparticle construction. This new approach paves the way for the development of antibody-targeted nanomedicines with improved paratope availability, reproducibility and uniformity to enhance both biological activity and ease of manufacture.
2017
50. Enabling the facile conversion of acyl hydrazides into N-acyl carbamates via metal-free ionic-based rupture of the N–N linkage
Shamsabadi, A., Ren, J., Chudasama, V.*
RSC Adv, 2017, 7, 27608-27611.
AbstractHerein we report the one-pot transformation of readily synthesised/easily accessed acyl hydrazides into N-acyl carbamates via an alkylation-elimination reaction sequence. A range of N-acyl carbamates are prepared in consistent yields, including those featuring protecting groups and having alkyl & aryl N-acyl functionality. The reaction conditions also tolerate a wide variety of sensitive functional groups.
49. Use of a next generation maleimide in combination with THIOMAB™ antibody technology delivers a highly stable, potent and near homogeneous THIOMAB™ antibody-drug conjugate (TDC)
Nunes, J.P.M., Vassileva, V., Robinson, E., Morais, M., Smith, M.E.B., Pedley, R. B., Caddick, S., Baker, J.R., Chudasama, V.*
RSC Adv, 2017, 7, 24828-24832.
AbstractHerein we demonstrate that conjugation of a next generation maleimide (NGM) to engineered cysteines in a THIOMAB™ antibody delivers a THIOMAB™ antibody-drug conjugate (TDC) with a drug loading of ca. 2. This TDC is highly stable in blood serum conditions, selective and potent towards HER2 expressing cell lines and meets the current criteria for optimised antibody-drug conjugates (ADCs).
48. Optimisation of the dibromomaleimide (DBM) platform for native antibody conjugation by accelerated post-conjugation hydrolysis
Morais, M., Nunes, J.P.M., Karu, K., Forte, N., Benni, I., Smith, M.E.B., Caddick, S., Chudasama, V.*, Baker, J.R.
Org Biomol Chem, 2017, 15, 2947-2952.
AbstractDisulfide bridging offers a convenient approach to generate site-selective antibody conjugates from native antibodies. To optimise the reagents available to achieve this strategy, we describe here the use of dibromomaleimides designed to undergo accelerated post-conjugation hydrolysis. Conjugation and hydrolysis, which serve to ‘lock’ the conjugates as robustly stable maleamic acids, is achieved in just over 1 h. This dramatic acceleration is also shown to infer significant improvements in homogeneity, as demonstrated by mass spectrometry analysis.
47. Pyridazinediones deliver potent, stable, targeted and efficacious antibody–drug conjugates (ADCs) with a controlled loading of 4 drugs per antibody
Robinson E., Nunes, J.P.M., Vassileva V., Maruani, A., Nogueira J.C.F., Smith, M.E.B., Pedley, B., Caddick, S., Baker, J.R., Chudasama, V.*
RSC Adv, 2017, 7, 9073-9077.
AbstractHerein we report the use of pyridazinediones to functionalise the native solvent accessible interstrand disulfide bonds in trastuzumab with monomethyl auristatin E (MMAE). This method of conjugation delivers serum stable antibody–drug conjugates (ADCs) with a controlled drug loading of 4. Moreover, we demonstrate that the MMAE-bearing ADCs are potent, selective and efficacious against cancer cell lines in both in vitro and in vivo models.
Lee, M.T.W., Maruani, A., Richards, D.A., Baker, J.R., Caddick, S., Chudasama, V.*
Chem Sci, 2017, 8, 2056-2060.
AbstractThe generation of antibody conjugates with a loading of two modules is desirable for a host of reasons. Whilst certain antibody engineering approaches have been useful in the preparation of such constructs, a reliable method based on a native antibody scaffold without the use of enzymes or harsh oxidative conditions has hitherto not been achieved. The use of native antibodies has several advantages in terms of cost, practicality, accessibility, time and overall efficiency. Herein we present a novel, reliable method of furnishing antibody conjugates with a loading of two modules starting from a native antibody scaffold.
Shamsabadi, A. and Chudasama, V.*
Org Biomol Chem, 2017, 15, 17-33.
AbstractHerein a review on the methods for the formation and reaction of acyl hydrazides will be given. There is particular focus on the synthesis of acyl hydrazides from aldehyde precursors with examination of the various approaches (e.g. metal-based (rhodium, copper) and non-metal-based (aerobically- and photoorganocatalytically-initiated)) that have been used to achieve this. Finally, strategies to utilise acyl hydrazides for the formation of an array of useful entities will be detailed.
Richards, D. A., Maruani, A. and Chudasama, V.*
Chem Sci, 2017, 8, 63-77.
AbstractRecent advances in nanomedicine have shown that dramatic improvements in nanoparticle therapeutics and diagnostics can be achieved through the use of disease specific targeting ligands. Although immunoglobulins have successfully been employed for the generation of actively targeted nanoparticles, their use is often hampered by the suboptimal characteristics of the resulting complexes. Emerging data suggest that a switch in focus from full antibodies to antibody derived fragments could help to alleviate these problems and expand the potential of antibody–nanoparticle conjugates as biomedical tools. This review aims to highlight how antibody derived fragments have been utilised to overcome both fundamental and practical issues encountered during the design and application of antibody–targeted nanoparticles.
2016
Smoktunowicz, N., Platé, M., Stern, A. O., D’Antongiovanni, V., Robinson, E., Chudasama, V., Caddick, S., Scotton, C. J., Jarai G. and Chambers, R. C.
Oncotarget, 2016, 7, 65471-65484.

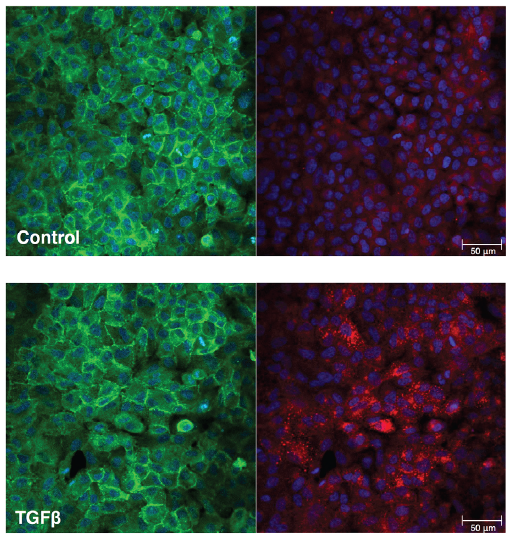
AbstractThe major high-affinity thrombin receptor, proteinase activated receptor-1 (PAR-1) is expressed at low levels by the normal epithelium but is upregulated in many types of cancer, including lung cancer. The thrombin-PAR-1 signalling axis contributes to the activation of latent TGFβ in response to tissue injury via an αvβ6 integrin-mediated mechanism. TGFβ is a pleiotropic cytokine that acts as a tumour suppressor in normal and dysplastic cells but switches into a tumour promoter in advanced tumours. In this study we demonstrate that TGFβ is a positive regulator of PAR-1 expression in A549 lung adenocarcinoma cells, which in turn increases the sensitivity of these cells to thrombin signalling. We further demonstrate that this effect is Smad3-, ERK1/2- and Sp1-dependent. We also show that TGFβ-mediated PAR-1 upregulation is accompanied by increased expression of integrin αv and β6 subunits. Finally, TGFβ pre-stimulation promotes increased migratory potential of A549 to thrombin. These data have important implications for our understanding of the interplay between coagulation and TGFβ signalling responses in lung cancer.
Pye, H., Butt M.A., Reinert, H.W., Maruani, A., Nunes, J.P.M., Marklew, J.S., Qurashi, M., Funnell, L., May, A., Stamati, I., Hamoudi, R., Baker, J.R., Smith, M.E.B., Caddick, S., Deonarain, M., Yahioglu, G., Chudasama, V.*, Lovat, L.
Photochem Photobiol Sci, 2016, 15, 1227-1238.
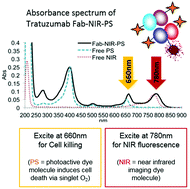
AbstractIn many cancers early intervention involves surgical resection of small localised tumour masses. Inadequate resection leads to recurrence whereas overzealous treatment can lead to organ damage. This work describes production of a HER2 targeting antibody Fab fragment dual conjugated to achieve both real time near-infrared fluorescent imaging and photodynamic therapy. The use of fluorescence emission from a NIR-dye could be used to guide resection of tumour bulk, for example during endoscopic diagnosis for oesophago-gastric adenocarcinoma, this would then be followed by activation of the photodynamic therapeutic agent to destroy untreated localised areas of cancer infiltration and tumour infiltrated lymph nodes. This theranostic agent was prepared from the Fab fragment of trastuzumab initially by functional disulfide re-bridging and site-specific click reaction of a NIR-dye. This was followed by further reaction with a novel pre-activated form of the photosensitiser chlorin e6 with the exposed fragments’ lysine residues. Specific binding of the theranostic agent was observed in vitro with a HER2 positive cell line and cellular near-infrared fluorescence was observed with flow cytometry. Specific photo-activity of the conjugates when exposed to laser light was observed with HER2 positive but not HER2 negative cell lines in vitro, this selectivity was not seen with the unconjugated drug. This theranostic agent demonstrates that two different photo-active functions can be coupled to the same antibody fragment with little interference to their independent activities.
Maruani, A., Richards, D.A., Chudasama, V.*
Org Biomol Chem, 2016, 14, 6165-6178.
AbstractWith the advent of novel bioorthogonal reactions and “click” chemistry, an increasing number of strategies for the single labelling of proteins and oligonucleotides have emerged. Whilst several methods exist for the site-selective introduction of a single chemical moiety, site-selective and bioorthogonal dual modification of biomolecules remains a challenge. The introduction of multiple modules enables a plethora of permutations and combinations and can generate a variety of bioconjuguates with many potential applications. From de novo approaches on oligomers to the post-translational functionalisation of proteins, this review will highlight the main strategies to dually modify biomolecules.
Chudasama, V.*, Maruani, A., Caddick, S.
Nature Chem, 2016, 8, 114-119.
AbstractAntibody–drug conjugates (ADCs) comprise antibodies covalently attached to highly potent drugs using a variety of conjugation technologies. As therapeutics, they combine the exquisite specificity of antibodies, enabling discrimination between healthy and diseased tissue, with the cell-killing ability of cytotoxic drugs. This powerful and exciting class of targeted therapy has shown considerable promise in the treatment of various cancers with two US Food and Drug Administration approved ADCs currently on the market (Adcetris and Kadcyla) and approximately 40 currently undergoing clinical evaluation. However, most of these ADCs exist as heterogeneous mixtures, which can result in a narrow therapeutic window and have major pharmacokinetic implications. In order for ADCs to deliver their full potential, sophisticated site-specific conjugation technologies to connect the drug to the antibody are vital. This Perspective discusses the strategies currently used for the site-specific construction of ADCs and appraises their merits and disadvantages.
Lee, M.T.W., Maruani, A., Baker, J.R., Caddick, S., Chudasama, V.*
Chem Sci, 2016, 7, 799-802.
AbstractHerein we present a significant step towards next-generation disulfide stapling reagents. A novel class of reagent has been designed to effect both disulfide reduction and functional re-bridging. The strategy has been applied to great success across various peptides and proteins. Moreover, application to a multi-disulfide system resulted in functional re-bridging without disulfide scrambling.
Gaitzsch, J., Chudasama, V.*, Morecroft, E., Messager, L., Battaglia, G.
ACS Macro Lett, 2016, 5, 351-354.
AbstractHerein we report the synthesis of an amphiphilic miktoarm star terpolymer and combine it with an equivalent diblock copolymer to form polymersomes with controlled surface topology. The three branches are ligated onto a central maleimide moiety in a reaction sequence that exploits various “click” chemistries. The final star was self-assembled with a linear block copolymer to generate a “patchy” surface on vesicles.
Lee, M.T.W., Maruani, A., Chudasama, V.*
J Chem Res, 2016, 40, 1-9.
AbstractThis article gives an overview of the use of 3,6-pyridazinediones in organic synthesis and chemical biology with an emphasis on recent developments. The properties of pyridazinediones, how they are constructed and how they have been applied in various fields of organic synthesis, medicinal chemistry and chemical biology will be highlighted.
Robinson, E., Knight, E., Smoktunowicz, N., Chambers, R., Inglis, G.G.A., Chudasama, V.*, Caddick S.
Org Biomol Chem, 2016, 14, 3198-3201.
AbstractThe discontinuation of PAR-1 antagonist RWJ-58259 beyond use as a biological probe is most likely due to it’s short half-life in vivo. However, retention of significant in vivo activity beyond the point where most of the RWJ-58259 had been consumed implies the generation of an active metabolite. Herein we describe the biological activity of a predicted metabolite of RWJ-58259 and the synthesis of analogues designed to enhance the metabolic stability of RWJ-58259.
Knight, E., Robinson, E., Smoktunowicz, N., Chambers, R., Aliev, A. E., Inglis, G.G.A., Chudasama, V.*, Caddick S.
Org Biomol Chem, 2016, 14, 3264-3274.
AbstractVorapaxar is a first-in-class PAR-1 antagonistic drug based on the ent-himbacine scaffold. Detailed in this article are enantioselective and racemic routes to various novel vorapaxar analogues. Biological testing revealed these compounds to have moderate to excellent potencies against PAR-1 with the most potent analogue demonstrating an IC50 of 27 nM.
Maruani, A., Lee, M.T.W, Watkins, G., Akhbar, A.R., Baggs, H., Shamsabadi, A
., Richards D.A.,
Chudasama, V.*
RSC Adv, 2016, 6, 3372-3376.
AbstractHerein we present an efficient method for the synthesis of esters from aromatic aldehydes via readily accessible acyl hydrazides. The developed reaction protocol is shown to be tolerant of a range of aromatic aldehydes, bearing various functionalities, as well as being amenable to the synthesis of thioesters and amides.
Richards D.A., Fletcher, S.A., Nobles, M., Kossen, H., Tedaldi, L., Chudasama, V., Tinker A., Baker, J.R.
Org Biomol Chem, 2016, 14, 455-459.
AbstractDescribed in this work is a novel method for photochemically manipulating peptides and proteins via the installation of cysteine-selective photoactive tags. Thiomaleimides, generated simply by the addition of bromomaleimides to reduced disulfide bonds, undergo [2 + 2] photocycloadditions to reconnect the crosslink between the two cysteine residues. This methodology is demonstrated to enable photoactivation of a peptide by macrocyclisation, and reconnection of the heavy and light chains in an antibody fragment to form thiol stable conjugates. Finally we report on an intriguing thiomaleimide mediated photochemical decarboxylation of C-terminal cysteines, discovered during this study.
2015
Maruani, A., Savoie, H., Bryden, F.,Caddick, S., Boyle, R.W., Chudasama, V.*
Chem Commun, 2015, 51, 15304-15307.
AbstractHerein we present a significant step towards next-generation antibody-based photodynamic therapeutics. Site-selective modification of a clinically relevant monoclonal antibody, with a serum-stable linker bearing a strained alkyne, allows for the controlled Cu-free “click” assembly of an in vitro active antibody-based PDT agent using a water soluble azide porpyhrin.
Smith, M.E.B., Caspersen, M.B., Robinson, E., Morais, M., Maruani, A., Nunes, J.P.M., Nicholls, K., Saxton, M.J., Caddick, S., Baker, J.R., Chudasama, V.*
Org Biomol Chem, 2015, 13, 7946-7949.
AbstractHerein we report the use of bromomaleimides for the construction of stable albumin conjugates via conjugation to its native, single accessible, cysteine followed by hydrolysis. Advantages over the classical maleimide approach are highlighted in terms of quantitative hydrolysis and absence of undesirable retro-Michael deconjugation.
Nunes, J.P.M., Morais, M., Vassileva V., Robinson E., Rajkumar, V., Smith, M.E.B., Pedley, B., Caddick, S., Baker, J.R., Chudasama, V.*
Chem Commun, 2015, 51, 10624-10627.
AbstractHerein we report the use of next generation maleimides (NGMs) for the construction of a potent antibody–drug conjugate (ADC) via functional disulfide bridging. The linker has excellent stability in blood serum and the ADC, armed with monomethyl auristatin E (MMAE), shows excellent potency and cancer cell selectivity in vitro.
Maruani, A., Smith, M.E.B., Miranda, E, Chester, K.A., Chudasama, V.*, Caddick, S.
Nature Commun, 2015, 6, 6645.
AbstractAlthough recent methods for the engineering of antibody–drug conjugates (ADCs) have gone some way to addressing the challenging issues of ADC construction, significant hurdles still remain. There is clear demand for the construction of novel ADC platforms that offer greater stability, homogeneity and flexibility. Here we describe a significant step towards a platform for next-generation antibody-based therapeutics by providing constructs that combine site-specific modification, exceptional versatility and high stability, with retention of antibody binding and structure post-modification. The relevance of the work in a biological context is also demonstrated in a cytotoxicity assay and a cell internalization study with HER2-positive and -negative breast cancer cell lines.
Chudasama, V.*
RSC Adv, 2015, 5, 44423-44426.
AbstractThe unique properties of pentafluorophenyl vinyl sulfonate allow for a hitherto unmet feat to be realised; efficient and high yielding, metal-free, radical-based alkene hydroacylation employing aldehyde as limiting reagent. The optimised conditions are shown to work in good yields across a series of aldehydes, thus demonstrating the wide applicability of the developed protocol.
Morgan, R.E., Chudasama, V., Moody, P., Smith, M.E.B., Caddick S.
Org Biomol Chem, 2015, 13, 4165-4168.
AbstractUbiquitination is of great importance as the post-translational modification of proteins with ubiquitin, or ubiquitin chains, facilitates a number of vital cellular processes. Herein we present a facile method of preparing various ubiquitin conjugates under mild conditions using michael acceptors based on dibromo-maleimides and dibromo-pyridazinediones.
Maruani, A., Alom, S., Canavelli, P., Lee, M., Morgan R.E., Chudasama, V.*, Caddick, S.
Chem Commun, 2015, 51, 5279-5282.
AbstractIt has recently emerged that the succinimide linkage of a maleimide thiol addition product is fragile, which is a major issue in fields where thiol functionalisation needs to be robust. Herein we deliver a strategy that generates selective cysteine thiol labelling reagents, which are stable to hydrolysis and thiol exchange.
2014
Schumacher, F.F., Nunes, J.P.M., Maruani, A., Chudasama, V., Smith, M.E.B., Chester, K.A., Baker, J.R., Caddick, S.
Org Biomol Chem, 2014, 7261-7269.
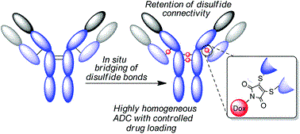
AbstractThe advent of Adcetris™ and Kadcyla™, two recently FDA-approved antibody–drug conjugates (ADCs), in the clinic has had a major impact on the treatment of lymphoma and breast cancer patients, respectively, worldwide. Despite these successes many new ADCs fail at various stages of development, often due to shortcomings in the methods used for their assembly. To address this problem we have developed next generation maleimides (NGMs), which specifically re-bridge reduced interchain disulfide bonds and allow the efficient conjugation of small molecules to antibodies, without the need for engineering of the target antibody. The method is site-specific and generates near homogeneous products in good yields. Moreover, adjustment of the reaction conditions allows control of the conjugation in terms of stoichiometry (drug-loading) and site selectivity. Using this method we prepared a series of ADCs from trastuzumab and doxorubicin (DOX) with a controlled drug-to-antibody ratio (DAR) of 1, 2, 3 and 4. All of these constructs were fully active by ELISA and had more than 90% of re-bridged disulfide bonds by CE-SDS when compared to clinical grade antibody. Furthermore, digest experiments of the DAR 2 material revealed that almost all of the drug had been targeted to the Fab arms of the antibody. Thus, NGMs offer a flexible and simple platform for the controlled assembly of ADCs from an antibody.
Moody, P.R., Chudasama, V., Nathani, R.I., Maruani, A., Martin, S., Smith, M.E.B., Caddick, S.
Chem Commun, 2014, 50, 4898-4900.
AbstractDesigned ankyrin repeat proteins (DARPins) are valuable tools in both biochemistry and medicine. Herein we describe a rapid, simple method for the dual modification of DARPins by introduction of cysteine mutations at specific positions that results in a vast difference in their thiol nucleophilicity, allowing for clean sequential modification.
Bryden, F., Maruani, A., Savoie, H., Chudasama, V., Smith, M.E.B., Caddick, S., Boyle, R.W.
Bioconjugate Chem, 2014, 25, 611-617.
AbstractThe rapidly increasing interest in the synthesis of antibody–drug conjugates as powerful targeted anticancer agents demonstrates the growing appreciation of the power of antibodies and antibody fragments as highly selective targeting moieties. This targeting ability is of particular interest in the area of photodynamic therapy, as the applicability of current clinical photosensitizers is limited by their relatively poor accumulation in target tissue in comparison to healthy tissue. Although synthesis of porphyrin–antibody conjugates has been previously demonstrated, existing work in this area has been hindered by the limitations of conventional antibody conjugation methods. This work describes the attachment of azide-functionalized, water-soluble porphyrins to a tratuzumab Fab fragment via a novel conjugation methodology. This method allows for the synthesis of a homogeneous product without the loss of structural stability associated with conventional methods of disulfide modification. Biological evaluation of the synthesized conjugates demonstrates excellent selectivity for a HER2 positive cell line over the control, with no dark toxicity observed in either case.
Browne, L.E., Nunes, J.P.M., Sim, J.A., Chudasama, V., Bragg, L., Caddick, S., North, R.A.
Proc Natl Acad Sci USA, 2014, 111, 521-526.
AbstractP2X receptors are trimeric membrane proteins that function as ion channels gated by extracellular ATP. We have engineered a P2X2 receptor that opens within milliseconds by irradiation at 440 nm, and rapidly closes at 360 nm. This requires bridging receptor subunits via covalent attachment of 4,4′-bis(maleimido)azobenzene to a cysteine residue (P329C) introduced into each second transmembrane domain. The cis–trans isomerization of the azobenzene pushes apart the outer ends of the transmembrane helices and opens the channel in a light-dependent manner. Light-activated channels exhibited similar unitary currents, rectification, calcium permeability, and dye uptake as P2X2 receptors activated by ATP. P2X3 receptors with an equivalent mutation (P320C) were also light sensitive after chemical modification. They showed typical rapid desensitization, and they could coassemble with native P2X2 subunits in pheochromocytoma cells to form light-activated heteromeric P2X2/3 receptors. A similar approach was used to open and close human acid-sensing ion channels (ASICs), which are also trimers but are unrelated in sequence to P2X receptors. The experiments indicate that the opening of the permeation pathway requires similar and substantial movements of the transmembrane helices in both P2X receptors and ASICs, and the method will allow precise optical control of P2X receptors or ASICs in intact tissues.
Akhbar, A.R., Chudasama, V.*, Fitzmaurice, R.J., Powell, L., Caddick, S.
Chem Commun, 2014, 50, 743-746.
AbstractIn this communication we describe a novel strategy for the formation of valuable diaryl and aryl alkyl ketones from acyl hydrazides. A wide variety of ketones are prepared and the mild reaction conditions allow for the use of a range of functionalities, especially in the synthesis of diaryl ketones.
Cal, P.M.S.D., Frade, R.F.M., Chudasama V., Cordeiro, C., Caddick, S., Gois, P.M.P.
Chem Commun, 2014, 50, 5261-5263.
AbstractHerein we present the synthesis of fluorescent 2-acetylbenzeneboronic acids that undergo B–N promoted conjugation with lysozyme and N-(2-aminoethyl) folic acid (EDA-FA), generating conjugates that are selectively recognized and internalized by cancer cells that over-express folic acid receptors.
Aliev A. E., Kulke, M., Khaneja, H. S,. Chudasama V., Sheppard, T. D., Lanigan R. M. Proteins, 2014, 82, 195-215.
AbstractWe propose a new approach for force field optimizations which aims at reproducing dynamics characteristics using biomolecular MD simulations, in addition to improved prediction of motionally averaged structural properties available from experiment. As the source of experimental data for dynamics fittings, we use 13C NMR spin-lattice relaxation times T1 of backbone and sidechain carbons, which allow to determine correlation times of both overall molecular and intramolecular motions. For structural fittings, we use motionally averaged experimental values of NMR J couplings. The proline residue and its derivative 4-hydroxyproline with relatively simple cyclic structure and sidechain dynamics were chosen for the assessment of the new approach in this work. Initially, grid search and simplexed MD simulations identified large number of parameter sets which fit equally well experimental J couplings. Using the Arrhenius-type relationship between the force constant and the correlation time, the available MD data for a series of parameter sets were analyzed to predict the value of the force constant that best reproduces experimental timescale of the sidechain dynamics. Verification of the new force-field (termed as AMBER99SB-ILDNP) against NMR J couplings and correlation times showed consistent and significant improvements compared to the original force field in reproducing both structural and dynamics properties. The results suggest that matching experimental timescales of motions together with motionally averaged characteristics is the valid approach for force field parameter optimization. Such a comprehensive approach is not restricted to cyclic residues and can be extended to other amino acid residues, as well as to the backbone.
18. SMITH, M., BAKER, J., CADDICK, S., CHUDASAMA, V. (2014). Thiol protecting group. Granted Patent EP2464654.
2013
Chudasama, V., Akhbar, A.R., Bahou, K.A., Fitzmaurice, R.J., Caddick, S.
Org Biomol Chem, 2013, 11, 7301-7317.
AbstractIn this report, a thorough evaluation of the use of aerobically initiated, metal-free hydroacylation of various CC and NN acceptor molecules with a wide range of aldehydes is presented. The aerobic-activation conditions that have been developed are in sharp contrast to previous conditions for hydroacylation, which tend to use transition metals, peroxides that require thermal or photochemical degradation, or N-heterocyclic carbenes. The mildness of the conditions enables a number of reactions involving sensitive reaction partners and, perhaps most significantly, allows for α-functionalised chiral aldehydes to undergo radical-based hydroacylation with complete retention of optical purity. We also demonstrate how the resulting hydroacylation products can be transformed into other useful intermediates, such as γ-keto-sulfonamides, sultams, sultones, cyclic N-sulfonyl imines and amides.
16. SMITH, M., BAKER, J., SCHUMACHER, F., CADDICK, S., CHUDASAMA, V., MARUANI, A. (2013). Chemical modification of antibodies. Patent application WO/2013/132268.
15. SMITH, M., BAKER, J., CADDICK, S., CHUDASAMA, V. (2013). Thiol protecting group. Granted Patent US13/389,625.
Castañeda, L., Maruani, A., Schumacher, F.F., Miranda, E., Chudasama, V., Chester, K.A., Baker, J.R., Smith, M.E.B., Caddick, S.
Chem Commun, 2013, 49, 8187-8189.
AbstractIn this communication we describe a novel acid-cleavable linker strategy for antibody–drug conjugation. Functional disulfide bridging of the single interchain disulfide bond of a trastuzumab Fab fragment yields a homogeneous antibody–drug conjugate bearing a thiomaleamic acid linker. This linker is stable at physiological pH and temperature, but quantitatively cleaves at lysosomal pH to release the drug payload.
Nathani, R.I., Moody, P.R., Chudasama, V., Smith M.E.B., Fitzmaurice, R.J., Caddick, S.
Chem Sci, 2013, 4, 3455-3458.
AbstractLocal protein microenvironment is used to control the outcome of reaction between cysteine residues and 2,5-dibromohexanediamide. The differential reactivity is exploited to introduce two orthogonal reactive handles onto the surface of a double cysteine mutant of superfolder green fluorescent protein in a regioselective manner. Subsequent elaboration with commonly used thiol and alkyne containing reagents affects site-selective protein dual labelling.
Castañeda, L., Wright, Z.V.F., Marculescu C., Tran, T.M., Chudasama V., Maruani, A., Hull E.A., Nunes J.P.M., Fitzmaurice R.J., Smith, M.E.B., Jones, L.H., Caddick, S., Baker, J.R. Tetrahedron Lett, 2013, 54, 3493-3495.
AbstractBromomaleimides are useful building blocks in synthesis and powerful reagents for the selective chemical modification of proteins. A mild new synthesis of these reagents is described, along with the convenient transferability of the approach to dithiomaleimides and bromopyridazinediones.
Nathani, R.I., Chudasama, V., Ryan, C.P., Moody P.R., Morgan, R.E., Fitzmaurice R.J., Smith, M.E.B., Baker, J., Caddick, S.
Org Biomol Chem, 2013, 2408-2411.
AbstractReversible protein biotinylation is readily affected via conjugation with a bromomaleimide-based reagent followed by reductive cleavage. The intermediate biotinylated protein constructs are stable at physiological temperature and pH 8.0. Quantitative reversibility is elegantly delivered under mild conditions of using a stoichiometric amount of a bis-thiol, thus providing an approach that will be of general interest in chemical biology and proteomics.
2012
Moody, P., Smith, M. E., Ryan, C. P., Chudasama, V., Baker, J. R., Molloy, J., Caddick, S.
Chembiochem, 2012, 13, 39-41.
AbstractBromomaleimides are versatile scaffolds that allow facile conjugation of thiolated biomolecules. Here we demonstrate that bromomaleimide-linked GFP–rhodamine FRET pairs cleave in the cytoplasm of mammalian cells. We believe that bromomaleimide scaffolds provide a potential core structure for prodrugs designed to release bioactive cargo following cell internalisation.
Brown, J. A., Chudasama, V., Giles M. E., Gill, D. M., Keegan, P. S., Kerr, W. J., Munday R. H., Griffin, K., Watts, A.
Org Biomol Chem, 2012, 10, 509-511.
AbstractTreatment of glycidyl sulfonamides with LDA delivers the corresponding enesulfonamide with good selectivity for the E-isomer, whereas the corresponding carbamates exhibit selectivity for the Z-enecarbamate. An E1cB elimination mechanism proceeding from a substrate–base chelate complex is advanced as rationalisation of the latter set of Z-selective outcomes.
2011
8. CADDICK, S., SMITH, M., BAKER, J., CHUDASAMA V. (2011). Thiol protecting group. Patent application WO/2011/018612
7. CADDICK, S., SMITH, M., BAKER, J., CHUDASAMA V. (2011). Reversible covalent linkage of functional moieties. Patent application WO 2011/018611
Chudasama, V., Ahern, J. M., Dhokia, D. V., Fitzmaurice, R. J., Caddick, S.
Chem Commun, 2011, 47, 3269-3271.
AbstractHerein we report the functionalisation of aldehydesvia hydroacylation of azodicarboxylates. A range of functionalised aldehydes are employed as the limiting reagent including chiral non-racemic aldehydes bearing α-stereocentres which are functionalised giving access to enantiomerically pure products. The resultant hydrazides can be employed as acyl donors in the synthesis of amides.
Chudasama, V., Ahern, J. M., Fitzmaurice, R. J., Caddick, S.
Tetrahedron Lett, 2011, 52, 1067-1069.
Abstractγ-Ketophosphonates are commonly employed as non-hydrolysable phosphate mimetics and as tools in synthesis. The synthesis of γ-ketophosphonates under mild conditions via interception of acyl radicals generated by aldehyde auto-oxidation is described.
Chudasama, V., Smith, M. E., Schumacher, F. F., Papaioannou, D., Waksman, G., Baker, J. R., Caddick, S.
Chem Commun, 2011, 47, 8781-8783.
AbstractBromopyridazinedione-mediated bioconjugation to a cysteine containing protein and a disulfide containing peptide is described. The conjugates are cleavable in an excess of thiol, including cytoplasmically-relevant concentrations of glutathione, and show a high level of hydrolytic stability. The constructs have the potential for four points of chemical attachment.
2010
Chudasama, V., Fitzmaurice, R. J., Ahern, J. M., Caddick, S.
Chem Commun, 2010, 46, 133-135.
AbstractHerein we report a mild, facile method for the preparation of 1,4-keto-sulfonates and sulfones onwater. Further synthetic manipulations can result in products that are not readily accessed by hydroacylation of electron rich alkenes.
Chudasama, V., Fitzmaurice, R. J., Caddick, S.
Nat Chem, 2010, 2, 592-596.
AbstractThe development of methods for carbon–carbon bond formation under benign conditions is an ongoing challenge for the synthetic chemist. In recent years there has been considerable interest in using selective C–H activation as a direct route for generating reactive intermediates. In this article, we describe the use of aldehyde auto-oxidation as a simple, clean and effective method for C–H activation, resulting in the generation of an acyl radical. This acyl radical can be used for carbon–carbon bond formation and herein we describe the application of this method for the hydroacylation of α,β-unsaturated esters without the requirement of additional catalysts or reagents. This methodology generates unsymmetrical ketones, which have been shown to have broad use in organic synthesis.
2008
Chudasama, V., Wilden J. D.
Chem Commun, 2008, 3768-3770.
AbstractA variety of five-membered ring oxazoles have been synthesised with complete regiocontrol and without the requirement for ring oxidationvia a reaction sequence based on a vinyl sulfonamide template.